
Chromosome-level genome and characteristic analysis of Platax teira
-
摘要:
尖翅燕鱼 (Platax teira) 具有生长速度快、肉质鲜美、营养价值高等特点,其外形奇特,尤其幼鱼更为特殊,可作为观赏鱼,是南海发展网箱养殖的重要潜力鱼类之一。由于目前缺乏尖翅燕鱼的基因组信息,其多数功能基因未被挖掘,已成为制约其遗传育种的重要因素。运用三代测序技术和组装获得染色体水平的尖翅燕鱼高质量基因组图谱,通过基因组注释获得尖翅燕鱼基因组序列的基本生物学信息。结果显示,尖翅燕鱼基因组大小为697.98 Mb,Hi-C挂载至24条染色体,挂载率为99.26%,含重复序列177.79 Mb,占整个基因组的25.47%,注释到22 851个蛋白编码基因。系统进化分析显示尖翅燕鱼与波纹唇鱼 (Cheilinus undulatus) 亲缘关系最近,分化时间距今约82.89 Ma。正选择基因主要富集在与离子通道和心脏功能有关的通路中,扩张的基因家族主要富集在嗅觉传导和氮代谢通路上,揭示了其在特定环境中的生存、适应依据和生态适应策略。
Abstract:Platax teira has the characteristics of fast growth rate, delicious meat and high nutritional content, and its strange appearance, especially for young fish, makes it an ornamental fish, being one of the important potential fishes for the cage culture development in the South China Sea. Due to the lack of genomic information, most of the functional genes of P. teira have not been explored, which has become an important factor of restricting its genetic breeding. We utilized triple sequencing technology and assembly to obtain a high-quality genome map of P. teira on chromosome level, and obtained the basic biological information of P. teira genome sequence through genome annotation. The results show that the genome size of P. teira was 697.98 Mb, assembling into 24 chromosomes with an assembly rate of 99.26%. P. teira genome contained 177.79 Mb of repetitive sequences, accounting for 25.47% of the total genome and encoding 22 851 genes. Comparative genomic analysis with 11 other fish species reveals that P. teira shared the closest relationship with Cheilinus undulatus, and the differentiation time was about 82.89 Ma. Genes under positive selection in P. teira were enriched in pathways related to ion channels and cardiac function, while the expanded gene families were enriched in pathways related to olfactory transmission and nitrogen metabolism, which reveals its survival, adaptation basis and ecological adaptation strategies in specific environment.
-
Keywords:
- Platax teira /
- Chromosome /
- Genome /
- Comparative genomics /
- Ephippidae
-
尖翅燕鱼 (Platax teira) 隶属于鲈形目、刺尾鱼亚目、白鲳科、燕鱼属,因为幼鱼的背鳍和腹鳍修长,又被称为长鳍蝙蝠[1-2]。其主要分布于印度-西太平洋区域,栖息于珊瑚礁潟湖区域,在中国分布在南海地区,主要以藻类、水母、浮游动物和小型底栖无脊椎动物为食[3]。尖翅燕鱼性情温顺、游动缓慢、生长快、肉质细嫩,是一种新兴的养殖种类[4],是南海发展网箱养殖的重要潜力鱼类之一。
国外主要开展了尖翅燕鱼生物地理学、行为学等方面的研究。Bilecenoglu和Kayam[5]、Golani等[3]研究发现了尖翅燕鱼种群在地中海地区的入侵和迁移;Leis等[6]研究了尖翅燕鱼的游泳能力。国内对尖翅燕鱼的研究主要集中于人工繁育和养殖。研究发现尖翅燕鱼生长主要受温度的影响[7],早期阶段生长速度较缓,后期生长迅速[8],通过投喂人工配合饲料,养殖5个月体质量可超0.5 kg[9],且不饱和脂肪酸含量丰富,具有较高的营养价值[10-13]。
近年来,随着测序技术的发展,基因测序取得了里程碑式的进展,很多物种都有了高质量的基因组图谱,并且基于基因组挖掘到了更多的功能基因,对于育种和生产起到不可替代的作用。然而,目前尚缺乏尖翅燕鱼的基因组信息,其育种养殖存在较大的瓶颈。本研究通过三代测序技术和组装获得染色体水平的尖翅燕鱼高质量基因组图谱,通过基因组注释获得尖翅燕鱼基因组序列的基本生物学信息,为后续深入开展尖翅燕鱼育种养殖及相关基因的研究奠定信息基础。
1. 材料与方法
1.1 样品采集与DNA提取
尖翅燕鱼实验样本采自海南陵水 (109°58'2.41''E, 18°24'51.46''N),样本经MS-222麻醉并用75% (φ) 的乙醇消毒鱼体。取尖翅燕鱼新鲜肌肉置于液氮中保存,用于Illumina、Pacbio和 Hi-C测序;取8个组织 (鳍、肠、肝、脾、鳃、肾、心、性腺) 置于液氮中保存,用于转录组测序和分析。
将尖翅燕鱼肌肉样本解冻,放入2 mL EP管中。剪碎,并加入560 μL DNA裂解液和4.5 μL RNA酶。用研磨棒对样本进行充分研磨以确保其完全裂解,同时用裂解液清洗研磨棒上的残留物。向样本中加入475 μL TE缓冲液和25 μL 10% (φ) 的SDS溶液,再加入10 μL 20 mg·mL–1蛋白酶K,将EP管放置于50 ℃的恒温水浴中消化2 h,期间每隔15 min颠倒混匀1次。加入等体积的酚∶氯仿∶异戊醇混合液,混合后4 ℃静置5 min,然后4 ℃、12 000 r·min–1离心10 min,将上清轻柔转移至新的2 mL EP管中。接着重复萃取过程以提高DNA纯度,随后加入2倍体积预冷的100% (φ)乙醇进行沉淀,轻轻颠倒混匀后 −20 ℃静置30 min。当出现白色沉淀时,用干净的小棒挑出放入新的EP管中,加入1.5 mL预冷的75% (φ) 乙醇,轻轻翻转后 −20 ℃静置5 min。再次挑出白色沉淀,晾干后加入35 μL无菌超纯水使DNA沉淀溶解。加入2 μL的10 mg·mL–1 RNA酶,在37 ℃水浴中孵育1 h后保存于 −20 ℃。最后通过0.8% (w) 琼脂糖凝胶电泳检测DNA的完整性,并利用Nanodrop分光光度计测定DNA浓度和纯度,以评估DNA的质量。
1.2 二代Illumina、三代PacBio、Hi-C测序
用限制性核酸内切酶,随机断裂尖翅燕鱼DNA,处理形成所需的片段,并通过加接头构建成片段文库。利用MGISEQ-2000平台分别进行双端 (Paired-End) 测序和单分子实时 (Single Molecule Real-Time) 测序,以获得全基因组的高通量测序数据,后续用于评估、组装和纠错基因组。
使用甲醛对尖翅燕鱼肌肉细胞进行交联处理,以固定染色质上的蛋白质与DNA之间的相互作用。利用限制性内切酶将固定后的DNA切割成许多较短的DNA片段。再将切割生成的DNA片段末端进行填充和修复,并在此步骤中加入生物素标记,以便于后续的富集和识别。之后将被切割和修复的DNA片段重新连接。由于交联,空间上接近的DNA片段更可能连接在一起,从而保留了它们之间的相互作用信息。去除甲醛交联,释放DNA,并通过生物素标记富集连接过的DNA片段,构建适用于高通量测序的DNA文库并进行测序。最后,通过Juicer-box和3D-DNA对测序数据进行分析,重建染色质之间的相互作用地图,揭示基因组的三维结构。
1.3 基因组数据质控、survey评估、基因组组装与Hi-C挂载
使用fastp软件的默认参数进行质控。fastp可自动对低质量序列和较多N的序列进行过滤,并自动化查找接头序列进行剪裁,还可对双端数据的碱基进行校正[14]。对三代测序得到的原始数据主要进行2个处理,去除低质量序列和接头序列。先利用SMRT Link[15]软件进行初始的数据质量评估,之后去除subreads与polymerase reads数据量的比例小于0.75和长度小于500 bp的reads。然后识别并去除序列中的接头,后续数据用于组装基因组。
对物种二代数据进行survey评估,应用k-mer分析法,本研究使用的k-mer数为17。组装采用NextDenovo和NextPolish方法。NextDenovo采取的是先矫正后组装的策略,能够在极大减少运行时间的情况下,达到甚至高于其他软件的纠错精度[16]。NextDenovo软件均设置为默认参数。用配套的NextPolish对得到的结果进行修正,参数设置与NextDenovo基本一致,最终得到高质量的基因组[17]。基因组完整度评估采用BUSCO法[18]。测得的二代数据用BWA的MEM模式比对出sam文件,用samtools的sort和flags查看二代测序和三代测序的匹配结果,用BUSCO查看基因组保守元件的完整性。
使用3D-DNA 和Juicer进行染色体水平组装[18]。先用BWA为基因组建索引,然后根据基因组创建可能的酶切位点文件,再提取每条contig的长度,最后运行Juicer得到3D-DNA输入文件。用3D-DNA得到Hi-C初步组装的草图,用Juicebox手动调整后再用3D-DNA得到最终染色体水平的单倍型基因组。
1.4 基因组重复序列注释、结构注释、功能注释
重复序列采用RepeatModeler和RepeatMasker进行注释。大致方法是:先使用RepeatModeler进行自我训练,构建尖翅燕鱼的重复序列文库;再用RepeatMasker加载Repbase数据库,并用 -species参数指定近缘种,参考RepeatModeler自我训练结果做重复序列预测[19],得出最终的重复序列。
基因组结构注释采取同源注释、转录组辅助注释、从头预测3种不同的方式进行,最终用EVM进行结果整合得到完整的结构注释结果[20]。选取黄金鲈 (Perca flavescens)、白吻梭鲈 (Sander lucioperca)、河鲈 (P. fluviatilis) 和橙喉镖鲈 (Etheostoma spectabile) 及模式物种斑马鱼 (Danio rerio) 5个物种进行同源注释。使用GeneWise软件对这些同源物种和尖翅燕鱼进行比对和基因预测,最终获得同源预测的基因集[21]。使用Hisat软件进行转录组辅助注释,将所有的转录本序列比对回尖翅燕鱼参考基因组上,使用Stringtie的merge参数进行多组织转录本序列的整合,再用p参数进行转录本预测,然后使用TransDecoder软件在预测的转录本中寻找存在开放阅读框的序列,得到有编码区 (CDS)的注释结果[22-23]。用上述2种注释方法结果文件做Augustus的训练集,通过隐马尔科夫模型训练具有完整结构的基因组集,这些结构包括基因的起始、终止位置和内含子剪接位点。然后利用这些基因集准确地从基因组中预测基因的结构[24]。最后使用EVM软件对3种注释方式的结果进行整合,设置3种预测结果权重,从头预测∶同源注释∶转录组辅助注释 (2∶5∶10),然后按软件默认参数正常运行得到整合结果。
功能注释主要通过序列比对的方法将得到的转录组结果与五大数据 (COG、KEGG、SwissProt、Trembl和NR) 进行序列比对,各数据库得到的注释最终汇总去冗余得到功能注释结果。
1.5 构建系统发育树及基因家族分析
将包括尖翅燕鱼在内的12个物种的基因组进行基因家族聚类,包括软骨鱼纲的大白鲨(Carcharodon carcharias),鲤形目的斑马鱼,鲈形目的泰国斗鱼 (Betta splendens)、眼斑双锯鱼 (Amphiprion ocellaris)、深裂眶锯雀鲷 (Stegastes partitus)、云纹石斑鱼 (Epinephelus moara)、波纹唇鱼 (Cheilinus undulatus),鳉形目的红箭鱼 (Xiphophorus helleri)、花斑剑尾鱼 (X. maculatus)、青鳉 (Oryzias latipes)、食蚊鱼 (Gambusia affinis)。选择依据为是否为珊瑚礁鱼类。用OrthoFinder对这些物种的蛋白序列聚类,统计得到单拷贝基因家族信息,用于构建系统进化树[25]。在系统发育树的基础上,借助PAML软件得到物种分化时间[26-28]。用CAFE 5分析尖翅燕鱼扩张收缩的基因家族[29]。用hmsearch在Pfam基因家族数据库中检索12个物种蛋白序列的结构域,将检索结果整合之后,过滤掉单个物种基因家族数目超过100的家族,以免影响CAFE 5的结果。然后将基因家族整合列表输入CAFE 5得到基因家族在各个物种和节点的扩张与收缩结果[30]。富集过程主要使用R软件包中的clusterProfiler进行,FDR<0.05且p<0.05的GO和KEGG通路被认为是显著富集的[31]。
2. 结果
2.1 基因组测序结果统计与Survey评估
采用Illumina测序平台对尖翅燕鱼的二代基因组进行双端测序,测序量为29.42 Gb;采用Pacbio测序平台对尖翅燕鱼进行三代基因组测序,测序量为88.1 Gb;对尖翅燕鱼进行Hi-C测序,获得数据量为13.85 Gb。这些数据均用于后续的基因组组装。
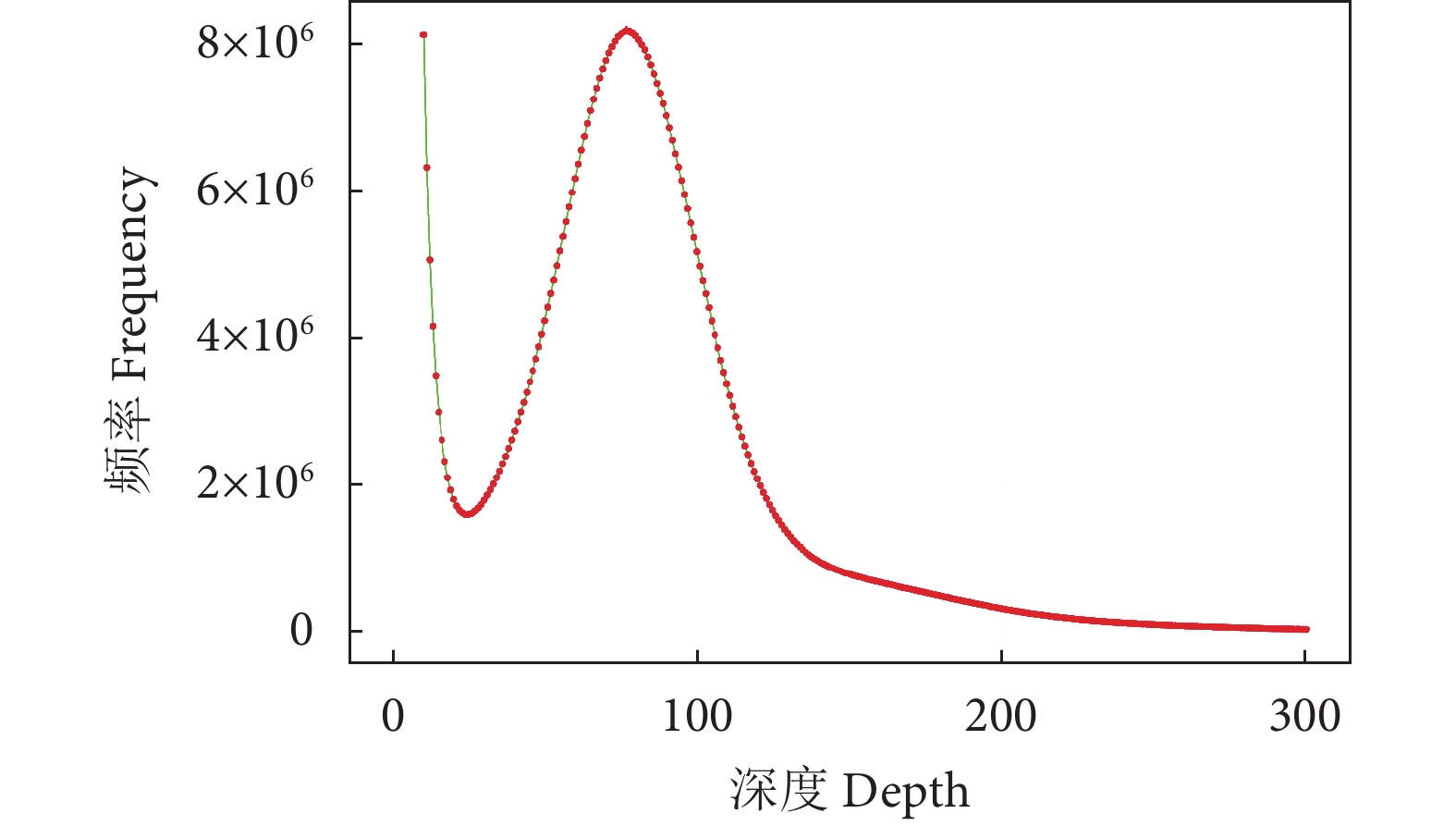
尖翅燕鱼的k-mer频数图仅有单峰 (图1),在k-mer深度为76时达到峰值,其为简单二倍体,组装策略比较简单。
2.2 二、三代基因组混合组装与Hi-C挂载结果统计
对Pacbio测序得到的数据过滤后进行组装,得到基因组大小为697.87 Mb的尖翅燕鱼数据,contig N50为26.17 Mb。使用NextPolish软件校正得到最终的基因组序列大小为697.98 Mb,contig N50为26.18 Mb,均有所提升,contig总数为134条。详细组装结果见表1。
表 1 尖翅燕鱼基因组组装和校正后组装结果统计Table 1. Statistical analysis of genome assembly and corrected assembly results of P. teira类型
TypeNextDenovo软件 NextPolish软件 长度
Length/bp数量
Count长度
Length/bp数量
CountN50 26 172 673 12 26 178 423 12 N90 8 653 897 29 8 656 320 29 最小长度
Min. length11 814 11 779 最大长度
Max. length33 665 020 33 669 695 平均长度
Ave. length5 207 983 5 208 839 总长度
Total length697 869 769 134 697 984 514 134 用BWA-MEM将二代和三代数据进行序列一致性评估,匹配率达99.59% (表2)。
表 2 尖翅燕鱼BWA比对结果Table 2. Results of BWA comparison of P. teira总读长
Total reads匹配读长
Map reads匹配率
Map rate/%双端读长
Paired reads双端比对读长
Paired map reads真正的读长
Proper paired reads正确匹配率
Properly map rate/%758 790 194 755 705 560 99.59 701 824 260 698 739 626 671 793 148 95.72 对组装的序列进行BUSCO预测,即用辐鳍鱼纲数据库 (actinopterygii_odb10) 中保守元件预测尖翅燕鱼的基因组组装情况,BUSCO完整性达到98%,说明绝大部分保守基因组装得比较完整,表明尖翅燕鱼基因组组装结果可信度较高 (表3)。
表 3 尖翅燕鱼基因组BUSCO评估Table 3. Genome BUSCO evaluation of P. teira类型
Type数量
Number百分比
Percentage/%完整的BUSCOs
Complete BUSCOs3 595 98.8 完整的单拷贝BUSCOs
Complete and single-copy BUSCOs3 571 98.1 完整的重复序列BUSCOs
Complete and duplicated BUSCOs24 0.7 碎片BUSCOs
Fragmented BUSCOs9 0.2 未比对上的BUSCOs
Missing BUSCOs36 1.0 总的BUSCO
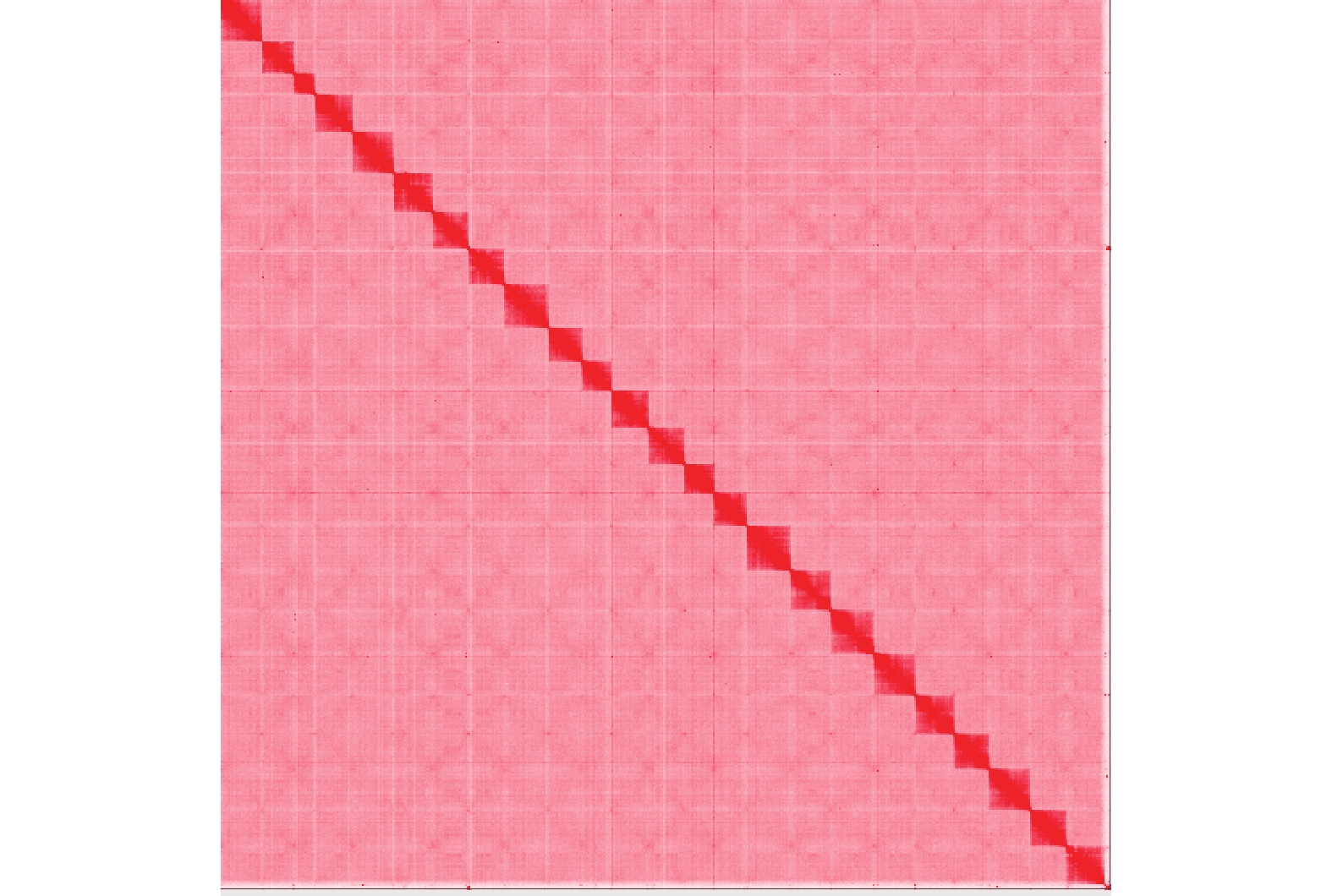
Total BUSCO3 640 100.0 经过3D-DNA聚类及Jucie-box手动调整,尖翅燕鱼染色体最终挂载到24条染色体上。挂载的碱基数692.84 Mb,占总序列的99.26%。最小的染色体仅有16.75 Mb,最大的染色体高达34.71 Mb (表4)。Hi-C交互热图很明显地区分为24条染色体 (图2),与尖翅燕鱼的核型分析相同 (2n=48)[32]。图2中红色越深代表交互作用越强,所有的染色体都是对角线位置的交互作用最强,说明Hi-C组装的染色体结果中邻近的染色体间交互强度高,与Hi-C辅助基因组组装的原理一致。在对角线以外区域未出现交互区,证明基因组挂载质量较高。
表 4 尖翅燕鱼各染色体长度Table 4. Length of each chromosome of P. teira染色体
Chromosome长度
Length/bp染色体
Chromosome长度
Length/bpHiC_scaffold_1 32 115 465 HiC_scaffold_13 22 937 044 HiC_scaffold_2 28 296 500 HiC_scaffold_14 29 051 425 HiC_scaffold_3 30 192 628 HiC_scaffold_15 28 201 383 HiC_scaffold_4 24 662 710 HiC_scaffold_16 23 075 928 HiC_scaffold_5 16 755 790 HiC_scaffold_17 25 711 309 HiC_scaffold_6 29 747 999 HiC_scaffold_18 34 711 268 HiC_scaffold_7 31 770 170 HiC_scaffold_19 31 217 840 HiC_scaffold_8 30 909 847 HiC_scaffold_20 34 223 832 HiC_scaffold_9 27 971 542 HiC_scaffold_21 32 267 482 HiC_scaffold_10 28 320 680 HiC_scaffold_22 30 609 981 HiC_scaffold_11 34 468 000 HiC_scaffold_23 27 290 755 HiC_scaffold_12 26 316 500 HiC_scaffold_24 32 023 099 合计 Total 692 849 177 2.3 基因组重复序列注释、结构注释、功能注释结果分析
共得到177 792 461 bp的重复序列,占整个基因组的25.47%,其中DNA转座子在注释出的元件中占比为7.94% (表5)。
表 5 尖翅燕鱼重复序列注释结果Table 5. Results of repeated sequence annotation of P. teira元件类型
Elements type元件数量
Number of
elements长度
Length/bp百分比
Percentage/%逆转录因子
Retroelements137 738 39 456 150 5.65 DNA转座子
DNA transposons266 011 55 441 011 7.94 环状
DNA Rolling-circles2 389 682 132 0.10 无分类
Unclassified366 703 66 929 139 9.59 总夹杂的重复序列
Total interspersed
repeats161 826 300 23.18 小RNA
Small RNA700 80 729 0.01 卫星DNA
Satellites DNA660 189 494 0.03 简单的重复序列
Simple repeats317 455 12 696 083 1.82 低复杂性
Low complexity42 640 2 317 723 0.33 总的重复
Total repeats177 792 461 25.47 EVM整合3种方法,最终预测出22 851个基因,基因长度平均为15 406.2 bp,CDS长度平均为172.26 bp,外显子个数平均为10.15,外显子长度平均为172.26 bp。
对基因预测结果进行BUSCO评估,与组装时的BUSCO评估类似,选用辐鳍鱼纲中保守的单拷贝同源基因评估基因组注释的完整性 (表6)。基因组中可以找到约88.3%的完整基因元件,相比于基因组BUSCO结果相差较大。
表 6 尖翅燕鱼结构注释结果BUSCO评估Table 6. BUSCO evaluation of structural annotation results of P. teira类型
Type数量
Number百分比
Percentage/%完整的BUSCOs
Complete BUSCOs3 213 88.3 完整的单拷贝BUSCOs
Complete and single-copy BUSCOs3 176 87.3 完整的重复序列BUSCOs
Complete and duplicated BUSCOs37 1.0 碎片BUSCOs
Fragmented BUSCOs155 4.3 未比对上的BUSCOs
Missing BUSCOs272 7.4 总的BUSCO
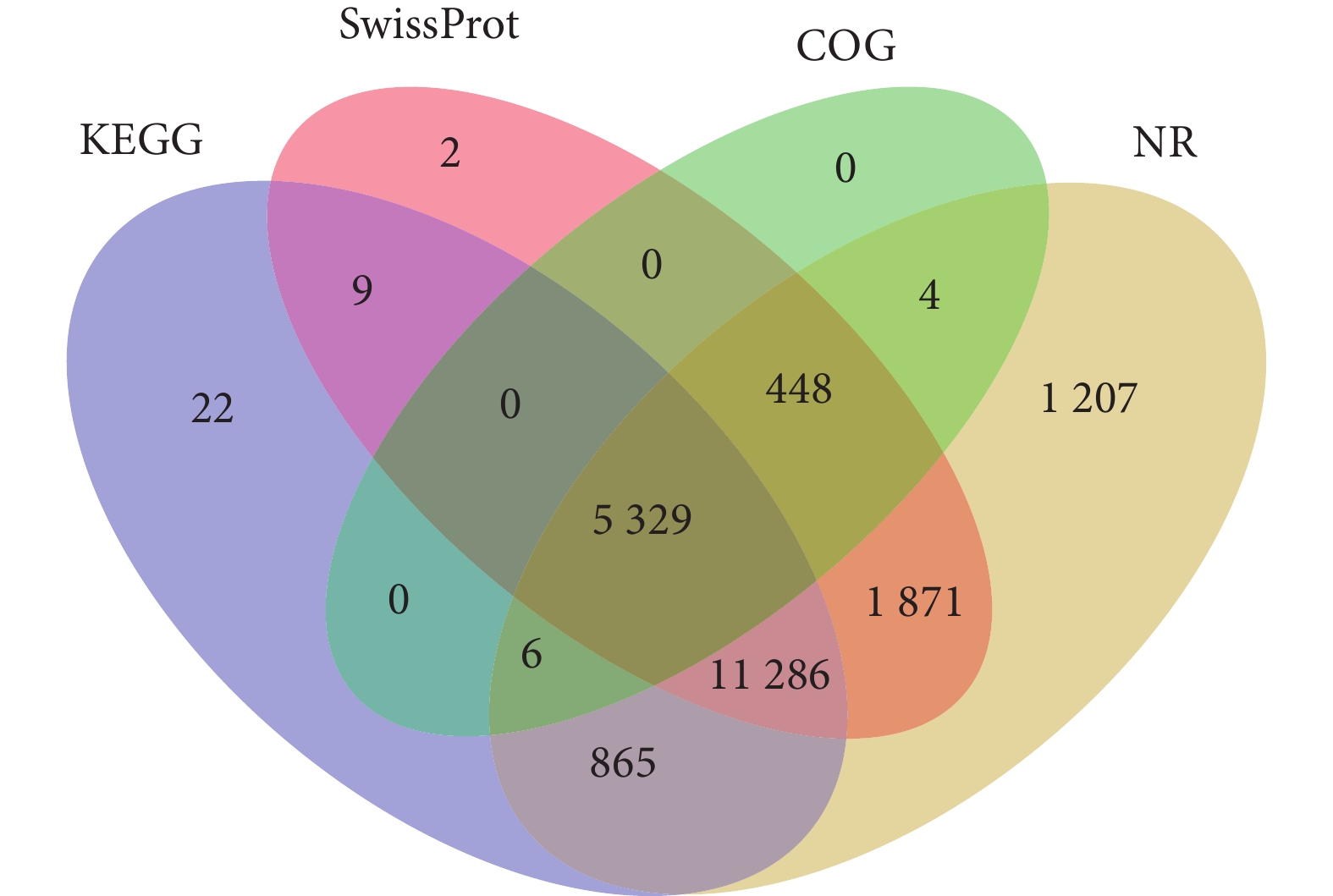
Total BUSCO3 640 100.0 对预测的22 851个基因进行功能注释,最终注释到功能的基因有21 104个,占比92.35% (表7)。
表 7 尖翅燕鱼功能注释结果Table 7. Functional annotation results of P. teira数据库
Database数量
Number百分比
Percentage/%NR 21 016 91.97 SwissProt 18 945 82.91 KEGG 17 511 76.63 COG 5 787 25.32 Trembl 20 996 91.88 功能注释基因
Functional annotation21 104 92.35 总计Total 22 851 100.00 几个数据库的功能注释结果重叠情况见图3。
![]() 图 3 尖翅燕鱼4个数据库注释结果韦恩图Figure 3. Venn diagram of four databases annotated results for P. teira
图 3 尖翅燕鱼4个数据库注释结果韦恩图Figure 3. Venn diagram of four databases annotated results for P. teira2.4 基因家族聚类
聚类分析共得到4 787个单拷贝基因。在选定的物种中斑马鱼的基因数目和特有基因数目最多,达32 717和3 503个,远比选定的其他物种多,一定程度上反映了其作为模式生物的复杂性和多样性。尖翅燕鱼的基因数目和大多数鱼类相近,但未聚类 (Unclustered) 的基因数目最多,达2 727个 (表8)。
表 8 12个物种基因家族聚类结果Table 8. Results of gene family clustering in 12 species物种
Species总的
Total单拷贝
Single特异性
Specific未聚类
Unclustered眼斑双锯鱼A. ocellaris 23 035 4 787 226 185 泰国斗鱼B. splendens 22 791 4 787 245 227 大白鲨C. carcharias 19 440 4 787 1 252 1 273 波纹唇鱼C .undulatus 23 316 4 787 381 184 斑马鱼D. rerio 32 717 4 787 3 503 1 659 云纹石斑鱼E .moara 23 735 4 787 112 336 食蚊鱼G. affinis 23 135 4 787 98 166 青鳉O. latipes 22 071 4 787 460 241 尖翅燕鱼P .teria 22 851 4 787 689 2 727 深裂眶锯雀鲷S. partitus 22 589 4 787 70 363 红箭鱼X. helleri 23 921 4 787 98 138 花斑剑尾鱼X. maculatus 23 238 4 787 34 126 2.5 系统发育树、物种分化时间估算
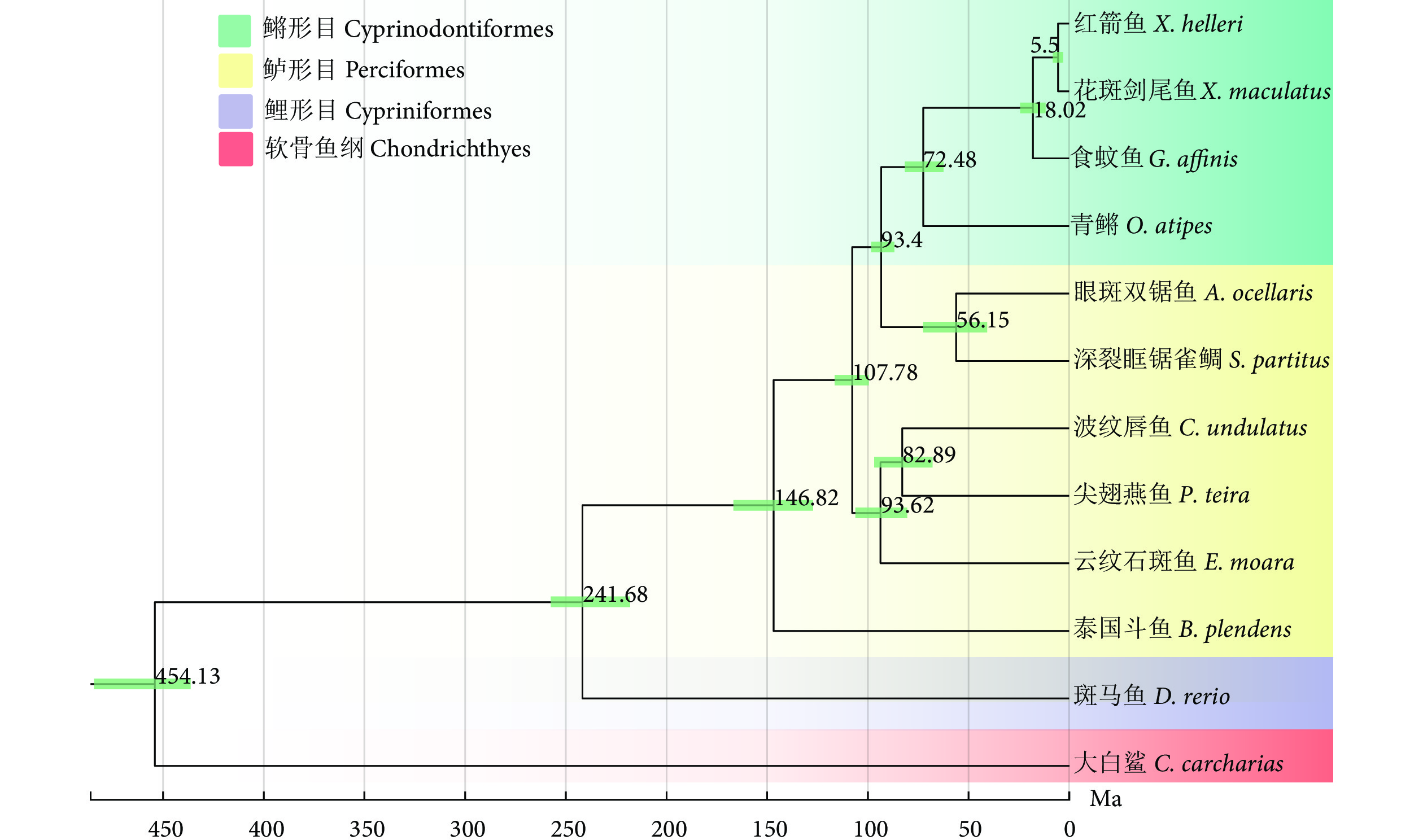
在系统发育树中,鳉形目的4个物种聚为一支,其中同属的红箭鱼和花斑剑尾鱼的亲缘关系最近。鲈形目雀鲷科的眼斑双锯鱼和深裂眶锯雀鲷聚为一支。鲈形目中体型较大的波纹唇鱼、云纹石斑鱼和尖翅燕鱼聚为一支,鲈形目的泰国斗鱼、鲤形目的斑马鱼、软骨鱼纲的大白鲨,形成3个单独的分支 (图4)。
分化时间估算结果表明,软骨鱼纲鱼类于距今约454.13 Ma与硬骨鱼纲鱼类发生分化,鲤形目与鲈形目和鳉形目的共同祖先在距今约241.68 Ma发生分化,鳉形目在距今约93.40 Ma与鲈形目产生分化。在图4中,尖翅燕鱼与波纹唇鱼亲缘关系最近,分化时间在距今约82.89 Ma。
2.6 基因家族的扩张和收缩结果
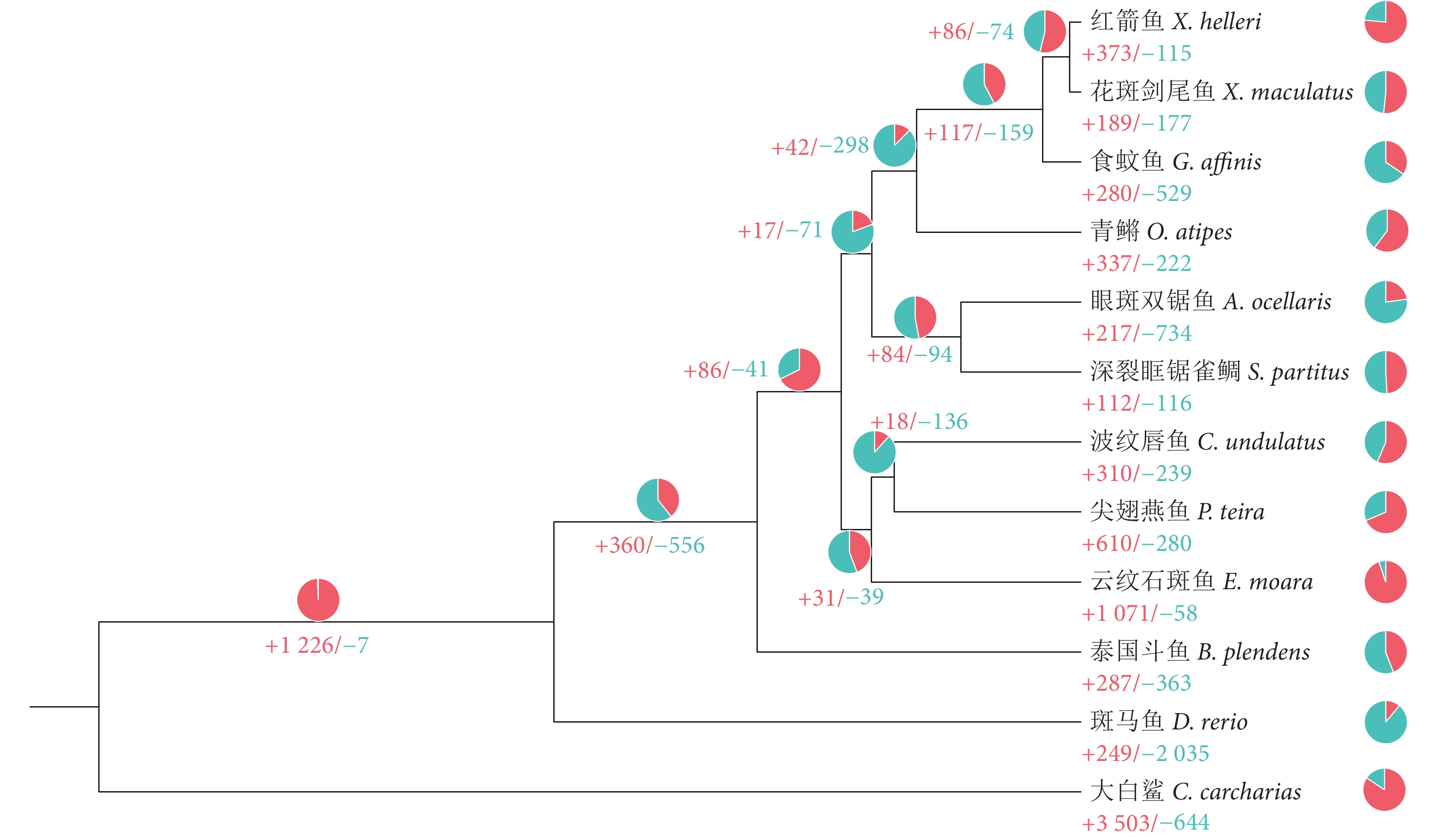
基因家族扩张与收缩分析结果显示,尖翅燕鱼扩张的基因家族有610个,收缩的基因家族有280个 (图5)。
![]() 图 5 各物种基因家族扩张收缩结果注:圈内红色代表扩张的基因家族;蓝色代表收缩的基因家族;物种名下方显示具体数值。Figure 5. Results of gene family expansion and contraction in each speciesNote: In the circle, red represents expanding gene families; blue represents contracting gene families,and specific values are displayed below the species name.
图 5 各物种基因家族扩张收缩结果注:圈内红色代表扩张的基因家族;蓝色代表收缩的基因家族;物种名下方显示具体数值。Figure 5. Results of gene family expansion and contraction in each speciesNote: In the circle, red represents expanding gene families; blue represents contracting gene families,and specific values are displayed below the species name.2.7 正选择基因分析
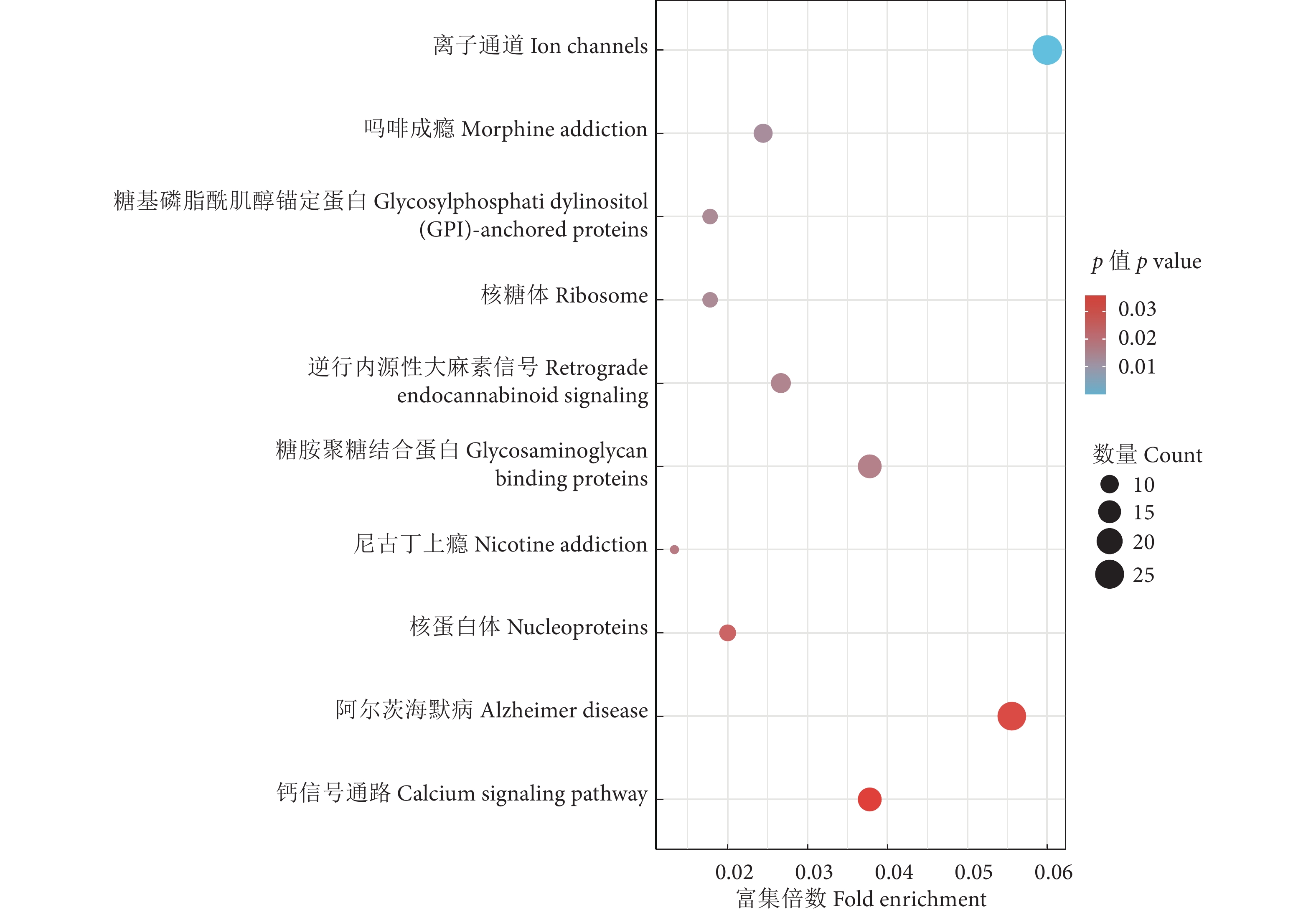
正选择分析结果显示,有803个单拷贝基因受到正选择。KEGG富集结果显示,尖翅燕鱼正选择基因主要富集在离子通道通路、吗啡成瘾通路、糖基磷脂酰肌醇 (GPI) 锚定蛋白的研究进展通路、核糖体通路、逆行内源性大麻素信号通路、糖胺聚糖结合蛋白通路、尼古丁上瘾通路、阿尔茨海默病和钙信号通路 (图6)。
![]() 图 6 尖翅燕鱼正选择基因KEGG富集结果Figure 6. KEGG pathway enrichment of positive selection genes identified in P. teira
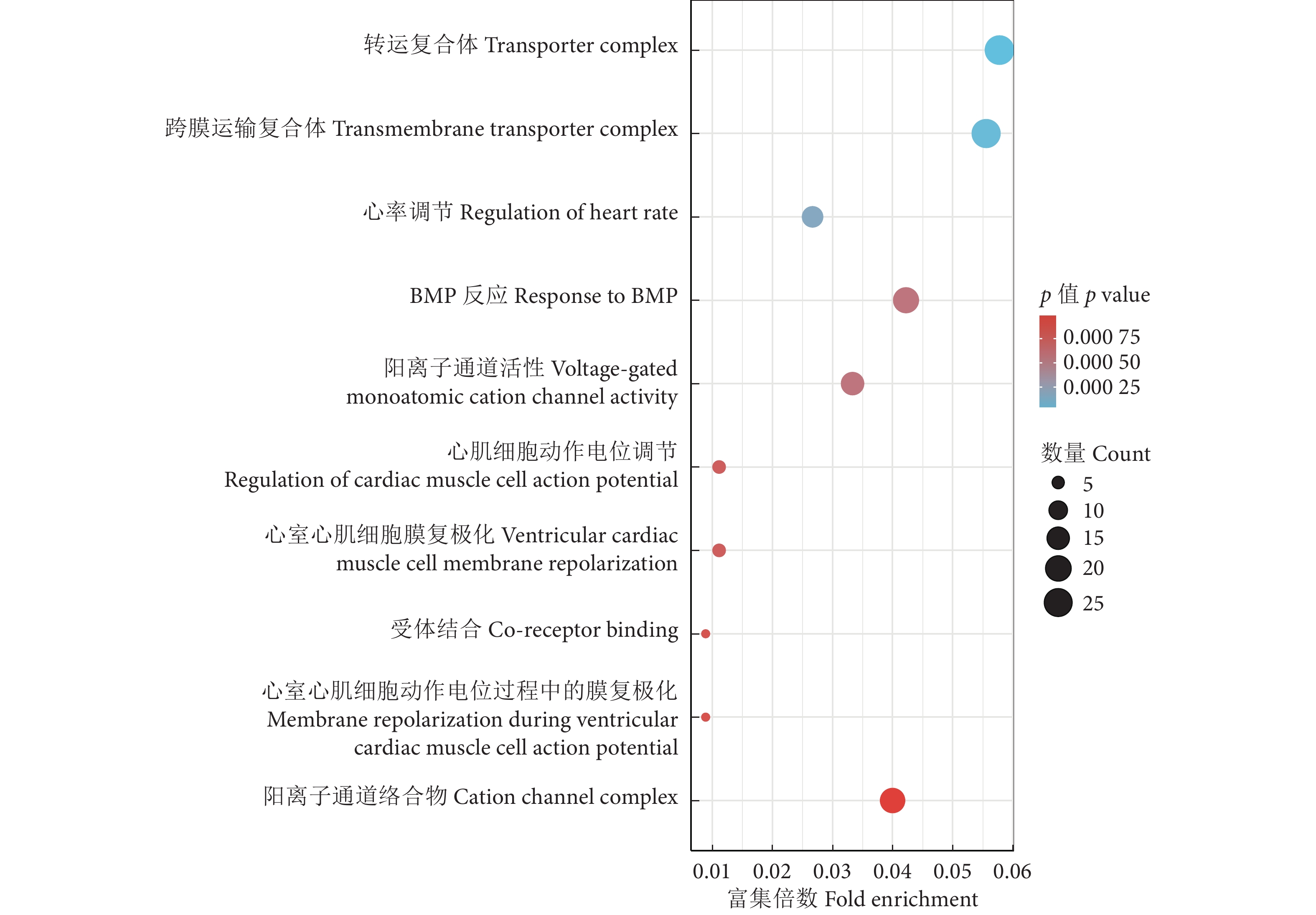
图 6 尖翅燕鱼正选择基因KEGG富集结果Figure 6. KEGG pathway enrichment of positive selection genes identified in P. teiraGO富集分析结果显示,尖翅燕鱼正选择基因主要富集在转运复合体、跨膜运输复合体、阳离子通道络合物等细胞组分,心率调节、BMP反应、心肌细胞动作电位的调节、心室心肌细胞膜复极化、心室心肌细胞动作电位过程中的膜复极化等生物过程,阳离子通道活性、受体结合等分子功能 (图7)。
![]() 图 7 尖翅燕鱼正选择基因GO富集结果Figure 7. GO enrichment of positive selection genes identified in P. teira
图 7 尖翅燕鱼正选择基因GO富集结果Figure 7. GO enrichment of positive selection genes identified in P. teira2.8 扩张基因家族富集分析
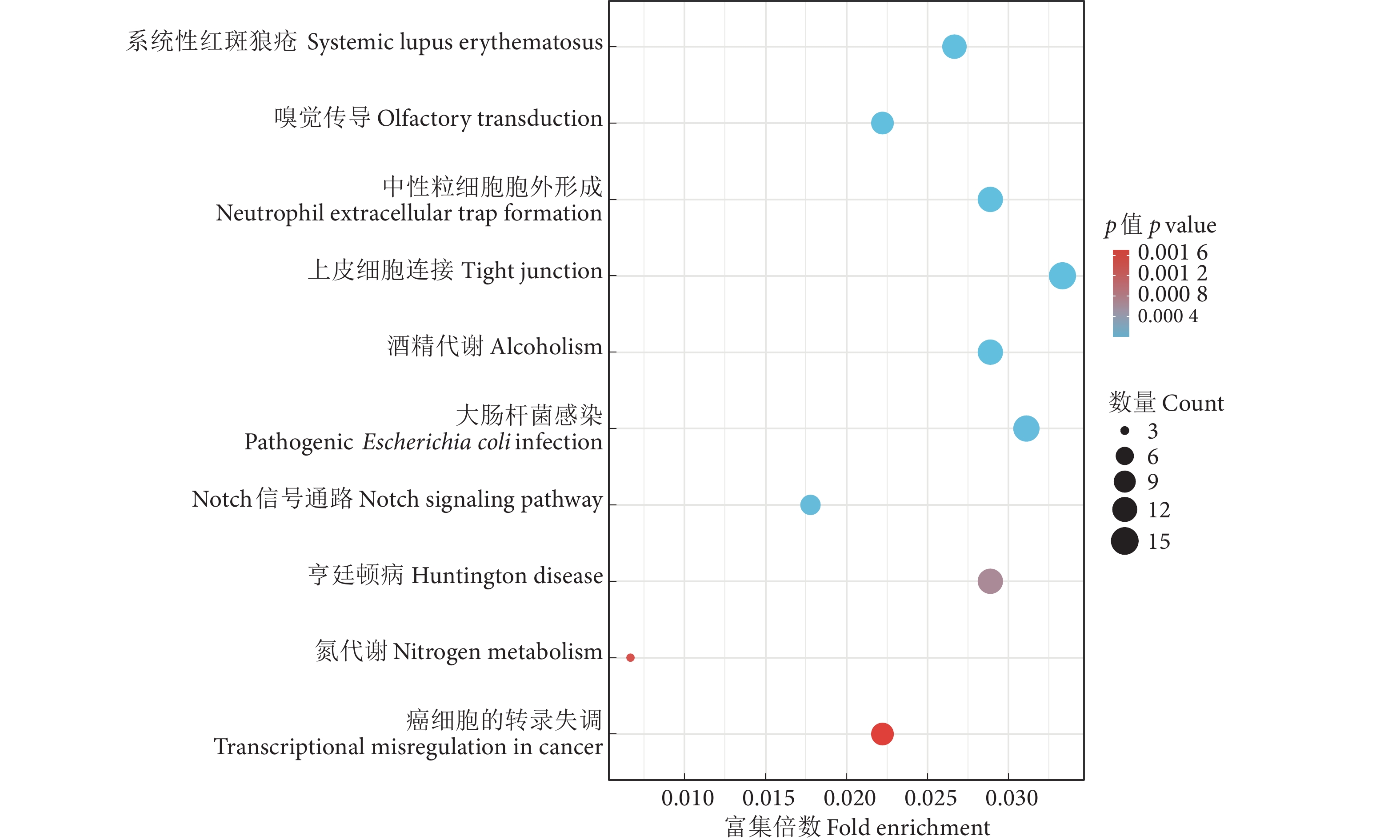
KEGG富集结果显示,尖翅燕鱼扩张的基因家族主要富集在系统性红斑狼疮、嗅觉传导、中性粒细胞胞外形成、上皮细胞连接、酒精代谢、大肠杆菌感染、Notch信号通路、亨廷顿病、氮代谢和癌细胞的转录失调通路 (图8)。
![]() 图 8 尖翅燕鱼扩张基因家族KEGG富集结果Figure 8. KEGG pathway enrichment of expand gene family identified in P. teira
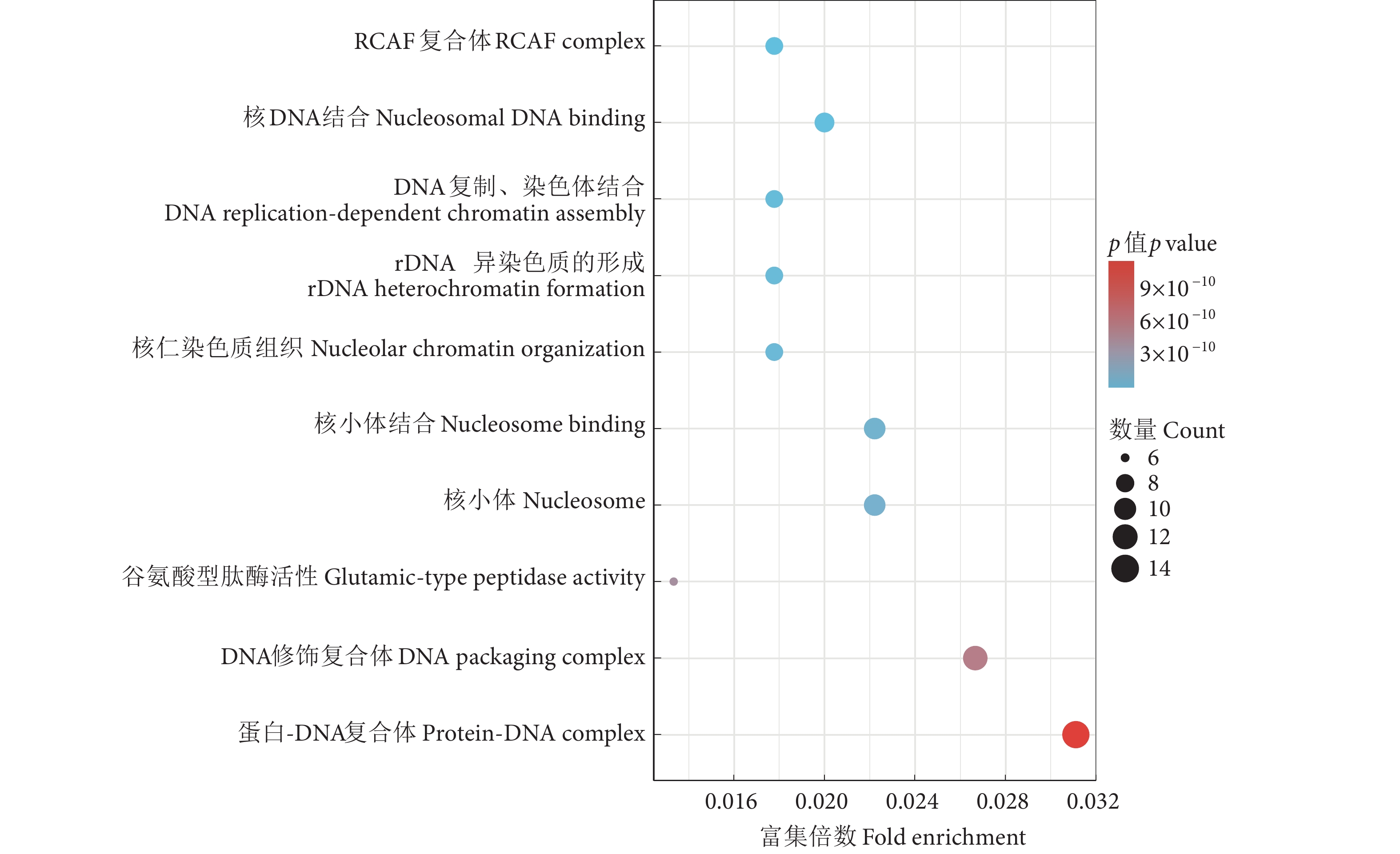
图 8 尖翅燕鱼扩张基因家族KEGG富集结果Figure 8. KEGG pathway enrichment of expand gene family identified in P. teiraGO富集分析结果显示,尖翅燕鱼扩张的基因家族主要富集在RCAF复合体、核小体、DNA修饰复合体、蛋白质-DNA复合体等细胞组分,核小体结合、DNA复制、染色质结合、谷氨酸型肽酶活性等分子功能,rDNA异染色质形成、核仁染色质组织、核小体结合等生物过程 (图9)。
![]() 图 9 尖翅燕鱼扩张基因家族GO富集结果Figure 9. GO pathway enrichment of expand gene family identified in P. teira
图 9 尖翅燕鱼扩张基因家族GO富集结果Figure 9. GO pathway enrichment of expand gene family identified in P. teira3. 讨论
硬骨鱼类也称为真骨鱼类,是鱼类中种类最多的一大类,包括了约96%的鱼类物种。硬骨鱼类的基因组大小也十分多样,最小的红鳍东方鲀 (Fugu rubripes) 基因组仅有300 Mb[33],而最大的中华鲟 (Acipenser sinensis) 基因组达8 Gb[34]。本研究从头组装得到尖翅燕鱼697.98 Mb大小的基因组,在鱼类中仅属于中等偏小。组装的N50达26 Mb,序列一致性评估达99.59%,BUSCO完整性达98%,均说明组装得到的基因组质量较高;Hi-C挂载到了24条染色体上,与尖翅燕鱼核型观察的染色体数目 (2n=48) 吻合,这个数目在经济鱼类中十分常见,如黄鳍棘鲷 (Acanthopagrus latus)[35]、卵形鲳鲹 (Trachinotus ovatus) 等[36]。挂载率达99.26%以及Hi-C互作图均反映出染色体级别的基因组组装效果良好,相比其他鱼类基因组组装的结果更好[37-43]。
鱼类基因组中的重复序列比例十分多样,大部分鱼类的基因组重复序列在30%左右,如大西洋鳕(Gadus morhua) 25.4%[44] 、三刺鱼 (Gasterosteus aculeatus) 25.2%[45]、大黄鱼 (Larimichthys crocea) 18.1%[46];在一些经历多倍体化事件的鱼类中,其基因组中的重复序列可能占到总基因组的一半以上,如斑马鱼[47]和大西洋鲑 (Salmo salar)[48]重复序列分别达52%和60%。基因组重复序列的注释显示,尖翅燕鱼基因组中重复序列占比26%,这个比例在鱼类中相对较低。这可能有助于尖翅燕鱼维持基因组的稳定性。尖翅燕鱼DNA 转座子在注释出的元件中占比7.94%,在已注释出的重复序列中占比最高,但相对于大多数鱼类20%左右的水平来说偏低[37-43]。值得注意的是,尚未注释的重复序列占全基因组的比例达9.59%,这些尚未注释的重复序列元件功能仍有待探知。
基因的结构注释最终获得22 851个基因,其中92.35%的基因均可在NR、SwissProt、KEGG、COG和InterPro等数据库中找到相应的功能信息。该注释结果为进一步的基因功能研究和通路分析提供了丰富的信息资源。
尖翅燕鱼在传统分类中被归类为白鲳科、燕鱼属。有学者基于白鲳科3个物种的线粒体基因组,对尖翅燕鱼及其近缘物种的系统发育关系进行了研究,发现线粒体基因组全长16 561 bp,由13个蛋白质编码基因、2个rRNA基因、22个tRNA基因和1个控制区组成[37]。本研究通过直系同源的单拷贝基因构建了12个物种带有分化时间的系统发育树,发现白鲳科的尖翅燕鱼与隆头鱼科的波纹唇鱼、石斑鱼科的云纹石斑鱼形成了一个单系类群,三者都是珊瑚礁鱼类中体型较大的类群,这可能表明大型的珊瑚礁鱼类存在较近的亲缘关系。同时,鳉形目的红箭鱼和花斑剑尾鱼、青鳉、食蚊鱼聚为一支,4个物种的共同祖先距今约72.48 Ma,其中食蚊鱼与剑尾鱼属在距今约18.02 Ma时期分化,红箭鱼和花斑剑尾鱼的分化时间距今约5.50 Ma,这些结果与Hughes等[42]构建的辐鳍鱼纲系统发育树结果相近。本研究结果为白鲳科的演化历史以及尖翅燕鱼在系统分类中的位置提供了新的参考。
研究发现正选择KEGG富集的结果主要集中在离子通道上,离子通道作为细胞感知和响应外部环境变化的最基本机制,在细胞信号传导和神经活动中扮演着关键角色,在鱼类的感知感觉系统中至关重要。在部分珊瑚礁鱼类中,感知感觉系统的特化存在惊人的相似。珊瑚礁生态的多彩促使许多珊瑚礁鱼类独立进化出了适应强烈光照和高清晰度视野的视觉能力。例如,小丑鱼和某些蝴蝶鱼都显示出对蓝色光的高度敏感[39]。在珊瑚礁鱼类中,特定的嗅觉受体家族同样在不同物种间显示出惊人的相似性。研究表明,如七鳃鳗科和鹦嘴鱼科的鱼类,尽管在进化树上位置相距甚远,但它们的嗅觉受体却表现出类似的基因表达模式和功能特性[49]。此外,钠、钾、钙离子通道还在鱼类的渗透压调节中起到关键作用,尼罗罗非鱼 (Oreochromis nilotica) 等广盐性鱼类可以通过离子通道来调整其体内的电解质平衡,以适应盐度变化的环境[50]。这种能力允许它们在不同的水域中迁移和生存,增加了它们的生态多样性和地理分布范围。离子通道的富集可能是尖翅燕鱼能在印度-太平洋区域以及地中海区域广泛分布并适应各种盐度环境的原因[1-2]。
利用KEGG和GO数据库对尖翅燕鱼的扩张基因家族进行富集分析发现,扩张的基因家族主要富集在嗅觉传导和氮代谢通路上。嗅觉传导通路是指从感知气味分子到大脑处理这些信息的一系列生物过程。这个过程涉及多个步骤,从气味分子的检测开始,经过信号转导,最终形成对气味的感知和行为反应。珊瑚礁鱼类通常具有高度发达的嗅觉系统,这使它们能够检测到极低浓度的水溶性化合物。这种嗅觉敏感性对于寻找食物、躲避捕食者、寻找配偶和定位适宜的栖息地至关重要,并在不同物种中具有趋同性,尤其是在繁殖期间或领域行为中。尖翅燕鱼有关嗅觉传导家族的显著扩张可能意味着其对环境和气味的高度感知能力,能使其在各种复杂的环境中更好地适应和生存。氮代谢通路主要包括氮的摄取、同化、转化、利用和排泄。在鱼类中氮的产生主要在肝脏线粒体基质中,可能通过氨转运蛋白将氨运输至鱼鳃处排泄[51]。对于大多数鱼类来说,氨是有毒的,内源性氨的积累会影响各种细胞过程[52]。氨通过激活胞质溶胶中的磷酸果糖激酶I来刺激鱼的糖酵解,还通过损害线粒体中的三羧酸循环来干扰能量代谢[53]。环境氨的积累则会导致鳃组织的病变[53]。事实上,在养殖水体中,饲料残饵、缺氧、鱼体死亡腐烂等各种因素都有可能导致养殖水体氨氮含量升高,氮代谢的显著富集预示着尖翅燕鱼可能存在高氨氮耐受能力,这也是其养殖的一大优势。
4. 结论
基于尖翅燕鱼的高质量基因组组装和注释以及比较基因组学分析,本研究成功地将尖翅燕鱼的基因组数据挂载至24条染色体上,并在基因组中识别出大量的重复序列和编码基因,提供了尖翅燕鱼的基因组信息和进化历史的数据。此外,通过与其他11种鱼类的比较基因组学分析发现,尖翅燕鱼与体型较大的珊瑚礁鱼类云纹石斑鱼和波纹唇鱼亲缘关系更近。尖翅燕鱼基因组中正选择基因的研究发现,基因功能主要富集在离子通道和心脏功能等关键生物通路上,为解析其在特定环境中的生存和适应提供了基因层面的依据。同时发现扩张的基因家族主要富集在嗅觉传导和氮代谢通路上,揭示了尖翅燕鱼的生态适应策略。
本研究结果丰富了白鲳科的基因组信息,也为功能基因组学研究和水产养殖业的发展提供了宝贵的资料,并为未来尖翅燕鱼的遗传改良和种群保护策略的制定提供了方向。
-
![]()
图 3 尖翅燕鱼4个数据库注释结果韦恩图
Figure 3. Venn diagram of four databases annotated results for P. teira
![]()
图 5 各物种基因家族扩张收缩结果
注:圈内红色代表扩张的基因家族;蓝色代表收缩的基因家族;物种名下方显示具体数值。
Figure 5. Results of gene family expansion and contraction in each species
Note: In the circle, red represents expanding gene families; blue represents contracting gene families,and specific values are displayed below the species name.
![]()
图 6 尖翅燕鱼正选择基因KEGG富集结果
Figure 6. KEGG pathway enrichment of positive selection genes identified in P. teira
![]()
图 7 尖翅燕鱼正选择基因GO富集结果
Figure 7. GO enrichment of positive selection genes identified in P. teira
![]()
图 8 尖翅燕鱼扩张基因家族KEGG富集结果
Figure 8. KEGG pathway enrichment of expand gene family identified in P. teira
![]()
图 9 尖翅燕鱼扩张基因家族GO富集结果
Figure 9. GO pathway enrichment of expand gene family identified in P. teira
表 1 尖翅燕鱼基因组组装和校正后组装结果统计
Table 1 Statistical analysis of genome assembly and corrected assembly results of P. teira
类型
TypeNextDenovo软件 NextPolish软件 长度
Length/bp数量
Count长度
Length/bp数量
CountN50 26 172 673 12 26 178 423 12 N90 8 653 897 29 8 656 320 29 最小长度
Min. length11 814 11 779 最大长度
Max. length33 665 020 33 669 695 平均长度
Ave. length5 207 983 5 208 839 总长度
Total length697 869 769 134 697 984 514 134  下载: 导出CSV
下载: 导出CSV
表 2 尖翅燕鱼BWA比对结果
Table 2 Results of BWA comparison of P. teira
总读长
Total reads匹配读长
Map reads匹配率
Map rate/%双端读长
Paired reads双端比对读长
Paired map reads真正的读长
Proper paired reads正确匹配率
Properly map rate/%758 790 194 755 705 560 99.59 701 824 260 698 739 626 671 793 148 95.72
下载: 导出CSV
表 3 尖翅燕鱼基因组BUSCO评估
Table 3 Genome BUSCO evaluation of P. teira
类型
Type数量
Number百分比
Percentage/%完整的BUSCOs
Complete BUSCOs3 595 98.8 完整的单拷贝BUSCOs
Complete and single-copy BUSCOs3 571 98.1 完整的重复序列BUSCOs
Complete and duplicated BUSCOs24 0.7 碎片BUSCOs
Fragmented BUSCOs9 0.2 未比对上的BUSCOs
Missing BUSCOs36 1.0 总的BUSCO
Total BUSCO3 640 100.0
下载: 导出CSV
表 4 尖翅燕鱼各染色体长度
Table 4 Length of each chromosome of P. teira
染色体
Chromosome长度
Length/bp染色体
Chromosome长度
Length/bpHiC_scaffold_1 32 115 465 HiC_scaffold_13 22 937 044 HiC_scaffold_2 28 296 500 HiC_scaffold_14 29 051 425 HiC_scaffold_3 30 192 628 HiC_scaffold_15 28 201 383 HiC_scaffold_4 24 662 710 HiC_scaffold_16 23 075 928 HiC_scaffold_5 16 755 790 HiC_scaffold_17 25 711 309 HiC_scaffold_6 29 747 999 HiC_scaffold_18 34 711 268 HiC_scaffold_7 31 770 170 HiC_scaffold_19 31 217 840 HiC_scaffold_8 30 909 847 HiC_scaffold_20 34 223 832 HiC_scaffold_9 27 971 542 HiC_scaffold_21 32 267 482 HiC_scaffold_10 28 320 680 HiC_scaffold_22 30 609 981 HiC_scaffold_11 34 468 000 HiC_scaffold_23 27 290 755 HiC_scaffold_12 26 316 500 HiC_scaffold_24 32 023 099 合计 Total 692 849 177
下载: 导出CSV
表 5 尖翅燕鱼重复序列注释结果
Table 5 Results of repeated sequence annotation of P. teira
元件类型
Elements type元件数量
Number of
elements长度
Length/bp百分比
Percentage/%逆转录因子
Retroelements137 738 39 456 150 5.65 DNA转座子
DNA transposons266 011 55 441 011 7.94 环状
DNA Rolling-circles2 389 682 132 0.10 无分类
Unclassified366 703 66 929 139 9.59 总夹杂的重复序列
Total interspersed
repeats161 826 300 23.18 小RNA
Small RNA700 80 729 0.01 卫星DNA
Satellites DNA660 189 494 0.03 简单的重复序列
Simple repeats317 455 12 696 083 1.82 低复杂性
Low complexity42 640 2 317 723 0.33 总的重复
Total repeats177 792 461 25.47
下载: 导出CSV
表 6 尖翅燕鱼结构注释结果BUSCO评估
Table 6 BUSCO evaluation of structural annotation results of P. teira
类型
Type数量
Number百分比
Percentage/%完整的BUSCOs
Complete BUSCOs3 213 88.3 完整的单拷贝BUSCOs
Complete and single-copy BUSCOs3 176 87.3 完整的重复序列BUSCOs
Complete and duplicated BUSCOs37 1.0 碎片BUSCOs
Fragmented BUSCOs155 4.3 未比对上的BUSCOs
Missing BUSCOs272 7.4 总的BUSCO
Total BUSCO3 640 100.0
下载: 导出CSV
表 7 尖翅燕鱼功能注释结果
Table 7 Functional annotation results of P. teira
数据库
Database数量
Number百分比
Percentage/%NR 21 016 91.97 SwissProt 18 945 82.91 KEGG 17 511 76.63 COG 5 787 25.32 Trembl 20 996 91.88 功能注释基因
Functional annotation21 104 92.35 总计Total 22 851 100.00
下载: 导出CSV
表 8 12个物种基因家族聚类结果
Table 8 Results of gene family clustering in 12 species
物种
Species总的
Total单拷贝
Single特异性
Specific未聚类
Unclustered眼斑双锯鱼A. ocellaris 23 035 4 787 226 185 泰国斗鱼B. splendens 22 791 4 787 245 227 大白鲨C. carcharias 19 440 4 787 1 252 1 273 波纹唇鱼C .undulatus 23 316 4 787 381 184 斑马鱼D. rerio 32 717 4 787 3 503 1 659 云纹石斑鱼E .moara 23 735 4 787 112 336 食蚊鱼G. affinis 23 135 4 787 98 166 青鳉O. latipes 22 071 4 787 460 241 尖翅燕鱼P .teria 22 851 4 787 689 2 727 深裂眶锯雀鲷S. partitus 22 589 4 787 70 363 红箭鱼X. helleri 23 921 4 787 98 138 花斑剑尾鱼X. maculatus 23 238 4 787 34 126
下载: 导出CSV
-
[1] BRAY R A, CRIBB T H. Lepocreadiidae (Digenea) from the batfish of the genus Platax Cuvier (Teleostei: Ephippidae) from the southern Great Barrier Reef, Queensland, Australia[J]. Syst Parasitol, 2003, 55(1): 1-9. doi: 10.1023/A:1023974022432
[2] MARIMUTHU N, WILSON J J, KUMARAGURU A K. Teira batrish, Platax teira (Forsskal, 1775) in Pudhumadam coastal waters, drifted due to the tsunami of 26 December 2004[J]. Current Sci, 2005, 89(8): 1310-1312.
[3] GOLANI D, SONIN O, EDELIST D. Second records of the Lessepsian fish migrants Priacanthus sagittarius and Platax teira and distribution extension of Tylerius spinosissimus in the Mediterranean[J]. Aquat Invasions, 2011, 6(S1): S7-S11.
[4] 刘明鉴, 郭华阳, 高杰, 等. 尖翅燕鱼早期胚胎发育及仔稚鱼形态观察[J]. 南方水产科学, 2022, 18(4): 103-111. doi: 10.12131/20210251 [5] BILECENOGLU M, KAYA M. A new alien fish in the Mediterranean Sea-Platax teira (Forsskål, 1775) (Osteichthyes: Ephippidae)[J]. Aquat Invasions, 2006, 1(2): 80-83. doi: 10.3391/ai.2006.1.2.5
[6] LEIS J M, HAY A C, HOWARTH G J. Ontogeny of in situ behaviours relevant to dispersal and population connectivity in larvae of coral-reef fishes[J]. Mar Ecol Prog Ser, 2009, 379: 163-179. doi: 10.3354/meps07904
[7] LIU M J, GAO J, GUO H Y, et al. Transcriptomics reveal the effects of breeding temperature on growth and metabolism in the early developmental stage of Platax teira[J]. Biology, 2023, 12(9): 1161. doi: 10.3390/biology12091161
[8] 陈松林, 徐文腾, 刘洋. 鱼类基因组研究十年回顾与展望[J]. 水产学报, 2019, 43(1): 1-14. [9] APARICIO S, CHAPMAN J, STUPKA E, et al. Whole-genomeshotgun assembly and analysis of the genome of Fugu rubripes[J]. Science, 2002, 297(5585): 1301-1310.
[10] LIU D, WANG X Y, GUO H Y, et al. Chromosome-level genome assembly of the endangered humphead wrasse Cheilinus undulatus: insight into the expansion of opsin genes in fishes[J]. Mol Ecol Resour, 2021, 21(7): 2388-2406.
[11] ZHENG S Q, SHAO F, TAO W J, et al. Chromosome-level assembly of southern catfish (Silurus meridionalis) provides insights into visual adaptation to nocturnal and benthic lifestyles[J]. Mol Ecol Resour, 2021, 21(5): 1575-1592. doi: 10.1111/1755-0998.13338
[12] 廖静. 人工养殖尖翅燕鱼性价比高[J]. 海洋与渔业, 2018(11): 62-63. [13] LIU B, GUO H Y, ZHU K C, et al. Nutritional compositions in different parts of muscle in the longfin batfish, Platax teira (Forsskål, 1775)[J]. J Appl Anim Res, 2019, 47(1): 403-407. doi: 10.1080/09712119.2019.1649680
[14] CHEN S F, ZHOU Y Q, CHEN Y R, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor[J]. Bioinformatics, 2018, 34(17): i884-i890. doi: 10.1093/bioinformatics/bty560
[15] ROBERTS R J, CARNEIRO M O, SCHATZ M C. The advantages of SMRT sequencing[J]. Genome Biol, 2013, 14(7): 405. doi: 10.1186/gb-2013-14-6-405
[16] HU J, WANG Z, SUN Z Y, et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads[J]. Genome Biol, 2024, 25(1): 107. doi: 10.1186/s13059-024-03252-4
[17] HU J, FAN J P, SUN Z Y, et al. NextPolish: a fast and efficient genome polishing tool for long-read assembly[J]. Bioinformatics, 2020, 36(7): 2253-2255. doi: 10.1093/bioinformatics/btz891
[18] SEPPEY M, MANNI M, ZDOBNOV E M. BUSCO: assessing genome assembly and annotation completeness[J]. Methods Mol Biol, 2019, 1962: 227-245.
[19] FLYNN J M, HUBLEY R, GOUBERT C, et al. RepeatModeler2 for automated genomic discovery of transposable element families[J]. PNAS, 2020, 117(17): 9451-9457. doi: 10.1073/pnas.1921046117
[20] HAAS B J, SALZBERG S L, ZHU W, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments[J]. Genome Biol, 2008, 9(1): R7. doi: 10.1186/gb-2008-9-1-r7
[21] BIRNEY E, CLAMP M, DURBIN R. GeneWise and Genomewise[J]. Genome Res, 2004, 14(5): 988-995. doi: 10.1101/gr.1865504
[22] KIM D, LANGMEAD B, SALZBERG S L. HISAT: a fast spliced aligner with low memory requirements[J]. Nat Methods, 2015, 12(4): 357-360. doi: 10.1038/nmeth.3317
[23] PERTEA M, PERTEA G M, ANTONESCU C M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads[J]. Nat Biotechnol, 2015, 33(3): 290-295. doi: 10.1038/nbt.3122
[24] STANKE M, DIEKHANS M, BAERTSCH R, et al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding[J]. Bioinformatics, 2008, 24(5): 637-644. doi: 10.1093/bioinformatics/btn013
[25] EMMS D M, KELLY S. OrthoFinder: phylogenetic orthology inference for comparative genomics[J]. Genome Biol, 2019, 20(1): 238. doi: 10.1186/s13059-019-1832-y
[26] KATOH K, STANDLEY D M. MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability[J]. Mol Biol Evol, 2013, 30(4): 772-780. doi: 10.1093/molbev/mst010
[27] KUMAR S, SULESKI M, CRAIG J M, et al. TimeTree 5: an expanded resource for species divergence times[J]. Mol Biol Evol, 2022, 39(8): msac174. doi: 10.1093/molbev/msac174
[28] YANG Z H. PAML 4: phylogenetic analysis by maximum likelihood[J]. Mol Biol Evol, 2007, 24(8): 1586-1591. doi: 10.1093/molbev/msm088
[29] MENDES F K, VANDERPOOL D, FULTON B, et al. CAFE 5 models variation in evolutionary rates among gene families[J]. Bioinformatics, 2021, 36(22/23): 5516-5518.
[30] EDDY S R. Accelerated profile HMM searches[J]. PLoS Comput Biol, 2011, 7(10): e1002195. doi: 10.1371/journal.pcbi.1002195
[31] YU G C, WANG L G, HAN Y Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters[J]. OMICS, 2012, 16(5): 284-287. doi: 10.1089/omi.2011.0118
[32] 高杰, 郭华阳, 刘明鉴, 等. 尖翅燕鱼染色体核型分析[J]. 海洋渔业, 2022, 44(5): 535-542. [33] OSHIUMI H , TSUJITA T , SHIDA K, et al. Prediction of the prototype of the human Toll-like receptor gene family from the pufferfish, Fugu rubripes, genome[J]. Immunogenetics, 2003, 54: 791-800.
[34] HU Y C, TAN R H, ZHU X, et al. Genome-wide identification, phylogeny and expressional profile of the Dmrt gene family in Chinese sturgeon (Acipenser sinensis)[J]. Sci Rep, 2024, 14(1): 4231. doi: 10.1038/s41598-024-54899-9
[35] ZHU K C, ZHANG N, LIU B S, et al. A chromosome-level genome assembly of the yellowfin seabream (Acanthopagrus latus; Hottuyn, 1782) provides insights into its osmoregulation and sex reversal[J]. Genomics, 2021, 113(4): 1617-1627. doi: 10.1016/j.ygeno.2021.04.017
[36] ZHANG D C, GUO L, GUO H Y, et al. Chromosome-level genome assembly of golden pompano (Trachinotus ovatus) in the family Carangidae[J]. Sci Data, 2019, 6(1): 216. doi: 10.1038/s41597-019-0238-8
[37] LIANG Y, XIAN L, PAN J M, et al. De Novo genome assembly of the whitespot parrotfish (Scarus forsteni): a valuable scaridae genomic resource[J]. Genes (Basel), 2024, 15(2): 249. doi: 10.3390/genes15020249
[38] ZHOU Q, GUO X Y, HUANG Y, et al. De novo sequencing and chromosomal-scale genome assembly of leopard coral grouper, Plectropomus leopardus[J]. Mol Ecol Resour, 2020, 20(5): 1403-1413. doi: 10.1111/1755-0998.13207
[39] CHEN X H, ZHONG L Q, BIAN C, et al. High-quality genome assembly of channel catfish, Ictalurus punctatus[J]. GigaScience, 2016, 5(1): 39. doi: 10.1186/s13742-016-0142-5
[40] LI J, BIAN C, HU Y C, et al. A chromosome-level genome assembly of the Asian arowana, Scleropages formosus[J]. Sci Data, 2016, 3: 160105.
[41] LI S S, XIE Z Z, CHEN P, et al. The complete mitochondrial genome of the Platax teira (Osteichthyes: Ephippidae)[J]. Mitochondrial DNA A DNA Mapp Seq Anal, 2016, 27(2): 796-797.
[42] HUGHES L C, ORTÍ G, HUANG Y, et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data[J]. PNAS, 2018, 115(24): 6249-6254. doi: 10.1073/pnas.1719358115
[43] HE S, LI L, LYU L Y, et al. Mandarin fish (Sinipercidae) genomes provide insights into innate predatory feeding[J]. Commun Biol, 2020, 3(1): 361. doi: 10.1038/s42003-020-1094-y
[44] MARSHALL H D, COULSON M W, CARR S M. Near neutrality, rate heterogeneity, and linkage govern mitochondrial genome evolution in Atlantic cod (Gadus morhua) and other gadine fish[J]. Mol Biol Evol, 2009, 26(3): 579-589.
[45] TEREKHANOVA N V, LOGACHEVA M D, PENIN A A, et al. Fast evolution from precast bricks: genomics of young freshwater populations of threespine stickleback Gasterosteus aculeatus[J]. PLoS Genet, 2014, 10(10): e1004696. doi: 10.1371/journal.pgen.1004696
[46] AO J Q, MU Y N, XIANG L X, et al. Genome sequencing of the perciform fish Larimichthys crocea provides insights into molecular and genetic mechanisms of stress adaptation[J]. PLoS Genet, 2015, 11(4): e1005118. doi: 10.1371/journal.pgen.1005118
[47] NIEDERRITER A R, DAVIS E E, GOLZIO C, et al. In vivo modeling of the morbid human genome using Danio rerio[J]. J Vis Exp, 2013(78): e50338.
[48] DAVIDSON W S, KOOP B F, JONES S J M, et al. Sequencing the genome of the Atlantic salmon (Salmo salar)[J]. Genome Biol, 2010, 11: 403. doi: 10.1186/gb-2010-11-9-403
[49] LEVANTI M, RANDAZZO B, VIÑA E, et al. Acid-sensing ion channels and transient-receptor potential ion channels in zebrafish taste buds[J]. Ann Anat, 2016, 207: 32-37. doi: 10.1016/j.aanat.2016.06.006
[50] MOHAMED N A, SAAD M F, SHUKRY M, et al. Physiological and ion changes of Nile tilapia (Oreochromis niloticus) under the effect of salinity stress[J]. Aquac Rep, 2021, 19: 100567. doi: 10.1016/j.aqrep.2020.100567
[51] IP Y K, CHEW S F. Ammonia production, excretion, toxicity, and defense in fish: a review[J]. Front Physiol, 2010, 1: 134.
[52] RANDALL D J, TSUI T K N. Ammonia toxicity in fish[J]. Mar Pollut Bull, 2002, 45(1/2/3/4/5/6/7/8/9/10/11/12): 17-23.
[53] ARILLO A, MARGIOCCO C, MELODIA F, et al. Ammonia toxicity mechanism in fish: studies on rainbow trout (Salmo gairdneri Rich.)[J]. Ecotoxicol Environ Saf, 1981, 5(3): 316-328. doi: 10.1016/0147-6513(81)90006-3
计量
- 文章访问数: 423
- HTML全文浏览量: 18
- PDF下载量: 22
 粤公网安备 44010502001741号
粤公网安备 44010502001741号
