
Determination and optimization of N-nitrosamines in salted fish by gas chromatography-mass spectrometry
-
摘要:
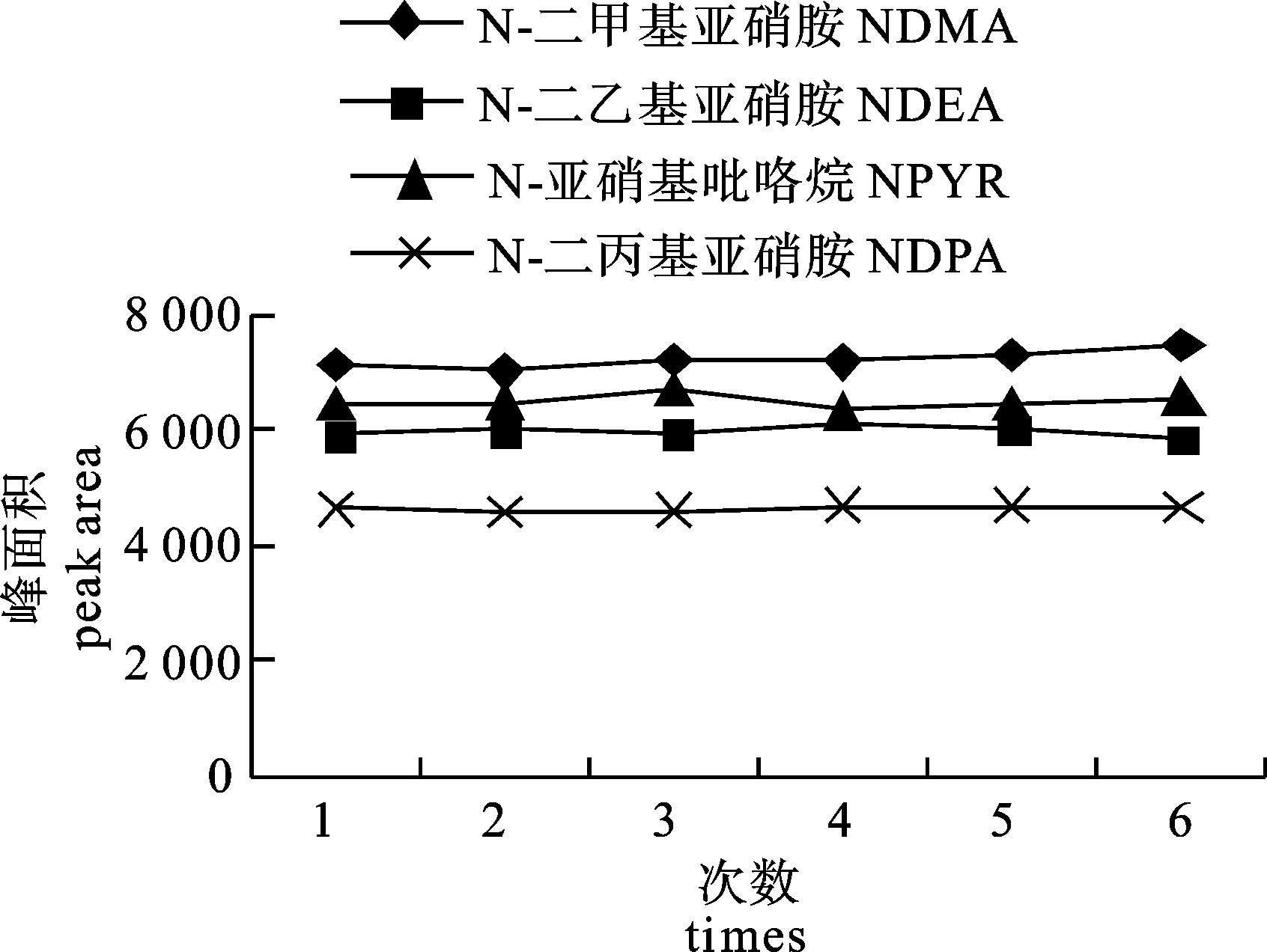
利用气相色谱-质谱联用(GC-MS)检测咸鱼中的N-亚硝胺,优化了样品前处理条件,比较了固相微萃取(SPME)和二氯甲烷超声萃取对N-亚硝胺的响应强度的影响,探讨了有机溶剂用量、萃取时间、萃取次数对测定的影响。采用选择离子法定性定量检测咸鱼中N-二甲基亚硝胺(NDMA)、N-二乙基亚硝胺(NDEA)、N-亚硝基吡咯烷(NPYR)和N-二丙基亚硝胺(NDPA)4种N-亚硝胺。结果显示,优化后的线性相关系数分别达到0.999 2、0.999 1、0.999 1和0.999 2;线性范围为0~10 μg·mL-1;该方法重现性好,其相对标准偏差(RSD)均≤2.1%;空白加标回收率可达70%~80%;灵敏度高,检测限分别为0.038 6 μg·kg-1、0.022 7 μg·kg-1、0.031 6 μg·kg-1和0.047 8 μg·kg-1。
Abstract:We determined N-nitrosamines in salted fish by gas chromatography-mass spectrometry (GC-MS), optimized the sample preparation conditions, compared the response intensity of N-nitrosamines by SPME and dichloromethane ultrasonic extraction as well as explored the impact of organic solvent, extraction time and times on the determination. By ion selective electrode method, we determined the contents of N-dimethylnitrosamine, N-diethylnitrosamine, N-nitroso-pyrrolidine and N-nitrosamines. Results show that the linear correlation coefficient is 0.999 2, 0.999 1, 0.999 1 and 0.999 2, respectively, with a linear range of 0~10 μg·mL-1. The method has good reproducibility (RSD≤2.1%, blank spiked recovery is 70%~80%) and high sensitivity (detection limit is 0.038 6 μg·kg-1, 0.022 7 μg·kg-1, 0.031 6 μg·kg-1 and 0.047 8 μg·kg-1, respectively).
-
Keywords:
- salted fish /
- N-nitrosamines /
- GC-MS /
- optimization
-
珠江由东江、西江和北江组成,生境异质性高,是目前生物多样性研究的热点地区之一[1]。西江全长2 214 km,流域面积为3.53×105 km2,年径流量达2 240×108 m3,孕育了丰富的鱼类资源[2]。然而,由于受到水利设施、环境污染、过度捕捞等因素的影响,西江鱼类资源退化严重[3-6],因此了解西江鱼类的遗传多样性现状和群体结构对今后鱼类资源利用与保护尤为必要。
鲫(Carassius auratus)属于鲤形目、鲤科、鲫属,为常见定居型鱼类,广泛分布于欧洲和亚洲诸多水系。由于其肉质细嫩鲜美,营养价值高,深受消费者喜爱,中国年消费量达200×104 t[7]。由于鲫具有较强的环境适应能力且易于被遗传改良,常被用于水产养殖[7]。鲫是珠江水系传统渔业的主要捕捞对象之一,但近几十年来其产量持续走低[1,8]。为了合理利用珠江鲫的种质资源,弄清该物种的遗传多样性现状和群体结构是关键。本研究利用线粒体细胞色素c氧化酶亚基Ⅰ (COⅠ)基因[9]作为分子标记[10],对西江水系鲫的8个地理群体共131尾样本开展遗传多样性和群体结构分析,旨在了解西江水系在高强度梯级开发、环境变化和人为干扰等条件下鲫的遗传多样性现状和群体结构,为鲫的资源利用和保护提供科学依据[11-12]。
1. 材料与方法
1.1 采样点设置
2016年在西江干流共设8个采样点,各采样点间平均间隔约200 km (图1和表1)。使用流刺网在8个采样站位共采集鲫样本131尾,剪取胸鳍或尾鳍保存于无水乙醇中,带回实验室后转移至 – 40 ℃冰箱内储存。
1.2 DNA的提取、扩增及测序
提取总DNA时,选取20 mg鳍条组织并将其铰成细小粒状,选用Axygen总基因组试剂盒进行提取,提取方法及步骤按照该试剂盒使用说明进行。使用通用引物(FishF1和FishR1)[13]对每尾样本进行PCR扩增。PCR反应体系为40 μL,10×buffer 4 μL,dNTPs (10 mmol·L–1) 2 μL,正反向引物各1 μL,Taq酶(5 U) 0.2 μL,基因组DNA 2 μL做反应模板,加超纯水补至40 μL。PCR反应条件为95 ℃预变性3 min;94 ℃变性30 s,54 ℃退火30 s,72 ℃延伸1 min,循环35次;最后72 ℃延伸10 min。扩增产物经1 %琼脂糖凝胶电泳检测后送至广州艾基生物技术有限公司进行双向测序,测序引物与扩增引物相同。
1.3 数据分析
使用Seqman软件对正反向测序结果进行拼接,运用Clustal X软件[14]对序列进行比对、编辑与剪切并辅以手工调整。使用MEGA 7.0 软件[15]计算遗传距离,模型选择Kimura双参数模型(K2P)。以鲤(Cyprinus carpio)作外类群,基于单倍型序列以邻接法(neighbor-joining,NJ)建树[16],使用1 000次重复抽样(bootstrap)检测其置信度,所用模型为K2P距离模型。使用Network 4.6软件[17]绘制单倍型网络图,并利用Dnasp 5.0软件 [18] 统计所有个体和每个群体的单倍型数,计算单倍型多样性(Hd)和核苷酸多样性(π)。使用Arlequin 3.5软件 [19],根据pairwise difference模型,估算两两群体间的遗传分化指数(FST),并基于Mantel检验分析地理距离是否与遗传分化存在联系。两两群体之间的地理距离大致通过采样点的经纬度坐标估算获得。本研究还对鲫群体进行分子方差分析(AMOVA),将干流的8个群体划分为一个组群,验证8个群体之间是否存在显著的遗传分化。最后,使用中性检验Fu's Fs和Tajima's D及核苷酸错配分布分析(mismatch distribution analysis)推断研究群体是否经历了近期的群体扩张。FST、AMOVA、中性检验和错配分布分析均采用1 000次重复。
2. 结果
本研究成功获得了131尾鲫的COⅠ基因片段。在序列两端截齐后,序列长度为636 bp,其中包含了36个变异位点,定义了25个单倍型。所有序列均未发现插入和缺失。邻接树的支持率较低,并未发现西江鲫形成了显著的谱系分枝(图2)。单倍型网络图表明,15个单倍型(H3~H9、H11、H12、H14~H18、H21)被多个地理群体共享。出现频率最高的单倍型是H4,占所有检测个体的26.0%,并且地理分布最广,为8个地理群体共享;其次出现频率较高的是H15,占总个体的10.7% (14/131)。有10个单倍型为单个地理群体所特有,其中,单倍型H1、H2仅在上游南盘江八渡江段分布;单倍型H10仅在中游红水河大化江段分布;单倍型H13等5个样本仅在中游红水河合山江段分布;单倍型H19、H20仅在下游西江藤县江段分布;单倍型H22、H23、H24和H25仅在下游西江肇庆江段分布。各单倍型网络关系分解见图3。
![]() 图 2 基于COⅠ基因构建的单倍型NJ系统发育树li1、li2和li3表示外类群Figure 2. Neighbor-Joining tree based on COⅠ geneThe li1, li2 and li3 represent the outgroups.
图 2 基于COⅠ基因构建的单倍型NJ系统发育树li1、li2和li3表示外类群Figure 2. Neighbor-Joining tree based on COⅠ geneThe li1, li2 and li3 represent the outgroups.![]() 图 3 基于COⅠ基因构建的鲫单倍型网络图圆面积代表单倍型的出现频率,彩色扇形面积代表各群体在同一单倍型中所占比例,空圈代表未发现或已灭绝的单倍型Figure 3. Median-joining haplotype network of C.auratus based on COⅠ geneCircle portion represents haplotype frequencies, while colored portion represents the proportions of the same haplotype that occurred in each sampling region. Empty circles represent haplotypes which were unfound or extinct.
图 3 基于COⅠ基因构建的鲫单倍型网络图圆面积代表单倍型的出现频率,彩色扇形面积代表各群体在同一单倍型中所占比例,空圈代表未发现或已灭绝的单倍型Figure 3. Median-joining haplotype network of C.auratus based on COⅠ geneCircle portion represents haplotype frequencies, while colored portion represents the proportions of the same haplotype that occurred in each sampling region. Empty circles represent haplotypes which were unfound or extinct.鲫总的单倍型多样性为0.900±0.017,核苷酸多样性为0.006 7±0.000 4,平均核苷酸差异数为4.231。各个地理群体的单倍型多样性介于0.714~0.917,核苷酸多样性介于0.001 4~0.008 7。此外,本研究发现上游站位的单倍型多样性和核苷酸多样性均低于中下游站位(表1)。
表 1 基于COⅠ基因序列的鲫群体遗传多样性参数Table 1. Genetic diversity ind in C.auratus populations based on COⅠ sequences群体
population经纬度
latitude and longitude样本数
numbr of samples单倍型数
number of haplotypes单倍型多样性指数 (Hd)
haplotype diversity核苷酸多样性指数 (π)
nucleotide diversity鲁布革 Lubuge 104°31′45″E, 24°44′46″N 17 5 0.809±0.057 0.002 8±0.000 7 八渡 Badu 105°48′26″E, 24°42′8″N 17 6 0.721±0.087 0.002 7±0.000 9 天峨 Tian'e 108°52′22″E, 23°48′43″N 7 3 0.714±0.127 0.001 4±0.000 3 大化 Dahua 107°59′16″E, 23°44′5″N 9 6 0.917±0.073 0.006 5±0.001 3 合山 Heshan 110°04′19″E, 23°24′16″N 19 8 0.836±0.057 0.008 7±0.000 9 桂平 Guiping 110°53′6″E, 23°21′46″N 21 8 0.833±0.055 0.005 8±0.001 1 藤县 Tengxian 112°27′33″E, 23°4′54″N 18 9 0.895±0.048 0.006 4±0.001 0 肇庆 Zhaoqing 110°04′20″E, 23°24′15″N 23 12 0.901±0.041 0.008 1±0.000 9 合计 total 131 25 0.900±0.017 0.006 7±0.000 4 AMOVA分析发现鲫群体间存在显著的遗传分化(FST=0.164, P=0.000),占总变异的16.40%(表2)。在估算两两群体之间的遗传分化系数时发现,鲁布革与大化、合山、藤县、肇庆群体间均存在显著的遗传分化(表3)。此外,八渡群体除与鲁布革群体未检测到显著遗传分化外,与其他江段群体都具有显著的遗传分化;藤县和肇庆2个群体与其他群体之间都有显著分化。但藤县和肇庆之间的FST为负值,说明两地理群体之间基因交流频繁。此外,藤县与距离较远的群体(鲁布革、八渡、天峨)之间的遗传分化值要明显大于距离较近的群体(大化、合山、桂平)。Mantel检验发现群体之间的地理距离与遗传分化呈现出显著的正相关关系(Mantel R=0.661,P=0.004,图4)。然而,部分距离较近的群体之间也存在显著的遗传分化,例如八渡与天峨之间,以及藤县与桂平之间。
表 2 基于COⅠ基因对珠江干流鲫的AMOVA分析Table 2. AMOVA analysis of C.auratus populations based on COⅠ gene变异来源
source of variation自由度
df平方和
SS变异组分
variance components变异百分数
percentage of variationFST P 群体间 among populations 7 52.692 0.354 46 Va 16.40 0.163 97 0.000 00 群体内 within populations 123 222.301 1.807 32 Vb 83.60 总计 total 130 274.992 2.161 78 表 3 基于COⅠ序列的野生鲫群体间的遗传分化系数 (FST)Table 3. FST analysis among populations of C.auratus based on COⅠ sequences群体
population鲁布革
Lubuge八渡
Badu天峨
Tian'e大化
Dahua合山
Heshan桂平
Guiping藤县
Tengxian鲁布革 Lubuge 八渡 Badu 0.010 27 天峨 Tian'e – 0.020 33 0.098 00* 大化 Dahua 0.124 63* 0.176 13* 0.099 99 合山 Heshan 0.123 91* 0.165 03* 0.118 59 0.061 15 桂平 Guiping 0.033 80 0.094 36* 0.043 29 0.081 67* 0.081 05* 藤县 Tengxian 0.279 76 * 0.319 31* 0.339 04* 0.227 87* 0.130 90* 0.144 19* 肇庆 Zhaoqing 0.279 36 * 0.313 49* 0.307 59* 0.190 04* 0.148 04* 0.150 29* – 0.019 77 注:*. P<0.05 ![]() 图 4 地理距离与两两群体间遗传分化的关系Figure 4. Relationship between geographic distance and pairwise differentiation
图 4 地理距离与两两群体间遗传分化的关系Figure 4. Relationship between geographic distance and pairwise differentiationTajima's D和Fu's Fs在总的群体和单个地理群体中均为不显著负值(表4),表明西江鲫群体可能并未经历近期的群体扩张。另外,错配分布分析未显示单峰分布(图5),并不支持群体历史上有过扩张。
表 4 基于COⅠ基因对野生鲫群体的中性检验Table 4. Neutrality test of C.auratus populations based on COⅠ gene群体
populationTajima's D Fu's Fs D P Fs P 鲁布革 Lubuge – 0.461 0.344 0.136 0.565 八渡 Badu – 1.550 0.054 – 1.005 0.227 天峨 Tian'e 0.206 0.598 – 0.237 0.241 大化 Dahua – 0.326 0.412 – 0.438 0.338 合山 Heshan 0.075 0.576 0.757 0.666 桂平 Guiping – 0.645 0.278 – 0.096 0.505 藤县 Tengxian – 0.838 0.205 – 1.042 0.318 肇庆 Zhaoqing – 0.785 0.244 – 1.869 0.226 总体 total – 0.540 0.339 – 0.474 0.386 ![]() 图 5 西江鲫群体核苷酸错配分布分析Figure 5. Mismatch distribution analysis of C.auratus in Xijiang River
图 5 西江鲫群体核苷酸错配分布分析Figure 5. Mismatch distribution analysis of C.auratus in Xijiang River3. 讨论
单倍型多样性(Hd)和核苷酸多样性(π)是衡量群体遗传多样性高低的重要指标[8]。诸多内外因素均会导致物种遗传多样性水平发生变化[20-23]。西江鲫总群体的Hd为0.900±0.017,π为0.006 7±0.000 4。依据Grant和Bowen[24]的标准,西江鲫的遗传多样性属于较高水平。袁振兴等[25]对线粒体DNA控制区 (D-loop)进行分析,发现贵州都柳江鲫的遗传多样性较高(Hd=0.892 6,π=0.006 32);邓朝阳[26]通过线粒体D-loop序列分析鲫的遗传多样性发现,分布于长江水系的野生鲫的遗传多样性同样较高(Hd =0.933,π=0.005 28)。另外一个有趣的发现是,鲁布革、八渡、天峨等群体的遗传多样性明显低于较下游的群体,大量的水利开发可能是导致这一结果的重要原因。由于大量的水电站建设,特别是中游红水河为十级梯级开发,已经将西江原河流截为多段,导致上游群体与中下游群体相对隔离,上游的鱼受泄洪及船闸排水的影响可迁移至下游江段,而中下游的鱼则难以迁移至较上游的区域,导致遗传多样性的差异分布[27]。
AMOVA分析和遗传分化系数(FST)估算表明西江鲫群体之间存在显著的遗传分化,而中下游群体具有的一些特有单倍型也从侧面反映出群体之间显著的遗传分化。FST估算发现距离较近的群体的遗传分化值往往小于距离远的群体,比如鲁布革群体与较近群体(八渡、天峨群体)之间的遗传分化值较小且不显著,而与相对较远的群体(鲁布革、天峨群体)之间的遗传分化值较高,达0.3。在Mantel检验中也发现群体之间遗传分化随着地理距离增大而显著增加。因此,地理距离是导致鲫遗传分化的一个重要因素。此外,本研究还发现部分距离较近的群体之间也存在显著的遗传分化,如藤县群体与桂平群体。自身的生活习性可能是导致距离较近群体间遗传分化的主要原因。鲫营定居性生活史,其迁徙能力有限,习惯生活于特定环境;而水利梯级开发将原河流截断,导致部分群体处于相对隔离的环境,限制了西江鲫群体之间的迁移扩散。
大量研究表明,周期性的冰川循环(glacial cycles)对许多现存物种的遗传多样性和分布都有重要的影响,但是不同物种或不同栖息环境的物种往往对冰期循环有着不同的响应[28-29]。本研究利用中性检验和错配分布分析了西江鲫的群体历史动态,发现近期西江鲫群体保持相对稳定的规模,并未经历群体扩张。这可能是由于西江地处热带和亚热带区域,在更新世冰期期间(glacial period)气候温和[30-31],鱼类能够维持其相对稳定的生态位,进而能保持相对稳定的群体数量。有研究表明,珠江流域的䱗(Hemiculter leucisculus)也经历了类似的群体历史动态[32]。
本研究探讨了西江鲫种群的遗传多样性现状和群体结构,结果显示鲫的遗传多样性处于相对较高的水平,地理距离和自身特定的生活史可能促进了鲫的遗传分化,西江鲫群体数量保持了相对稳定的状态。本研究结果为鲫的资源利用与保护以及今后珠江水系其他鱼类遗传多样性和群体结构的研究提供理论参考依据。由于本研究的分子标记单一且采样范围有限,因此后续研究有必要选取更多的分子标记(微卫星标记、简化基因组)并加大采样范围来进一步阐释鲫群体的遗传多样性现状和群体结构。
-
![]()
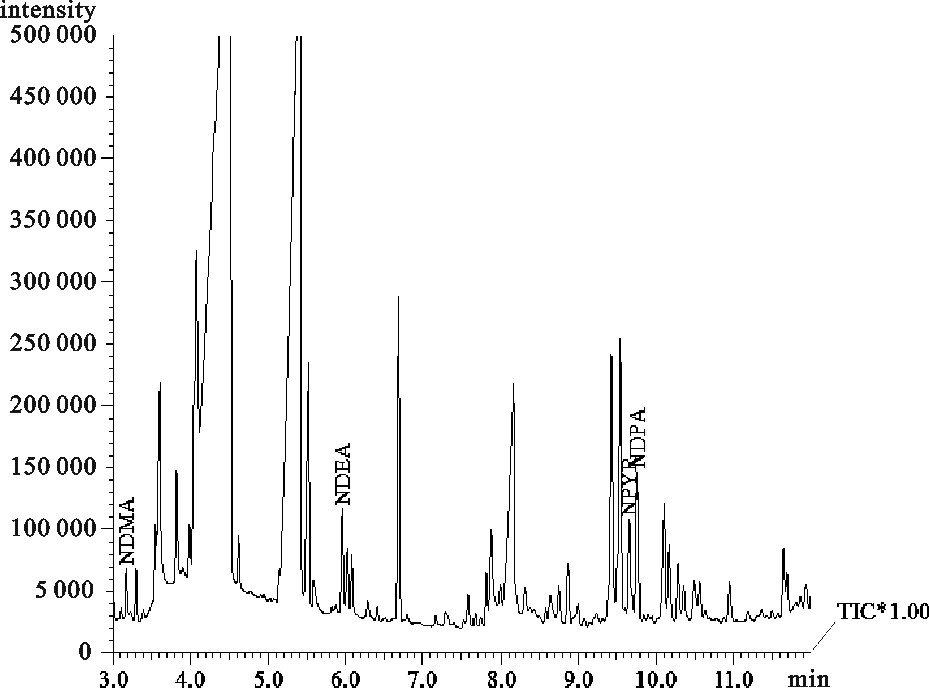
图 1 固相微萃取(a)和超声萃取(b)的色谱图
Figure 1. Chromatograms of SPME(a)and supersonic extraction(b)
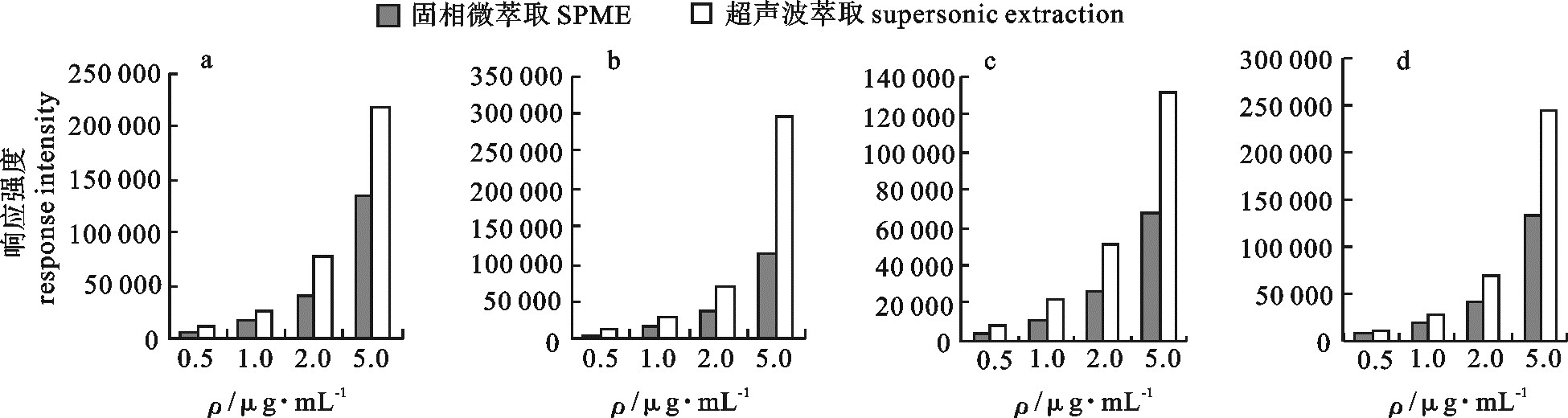
![]()
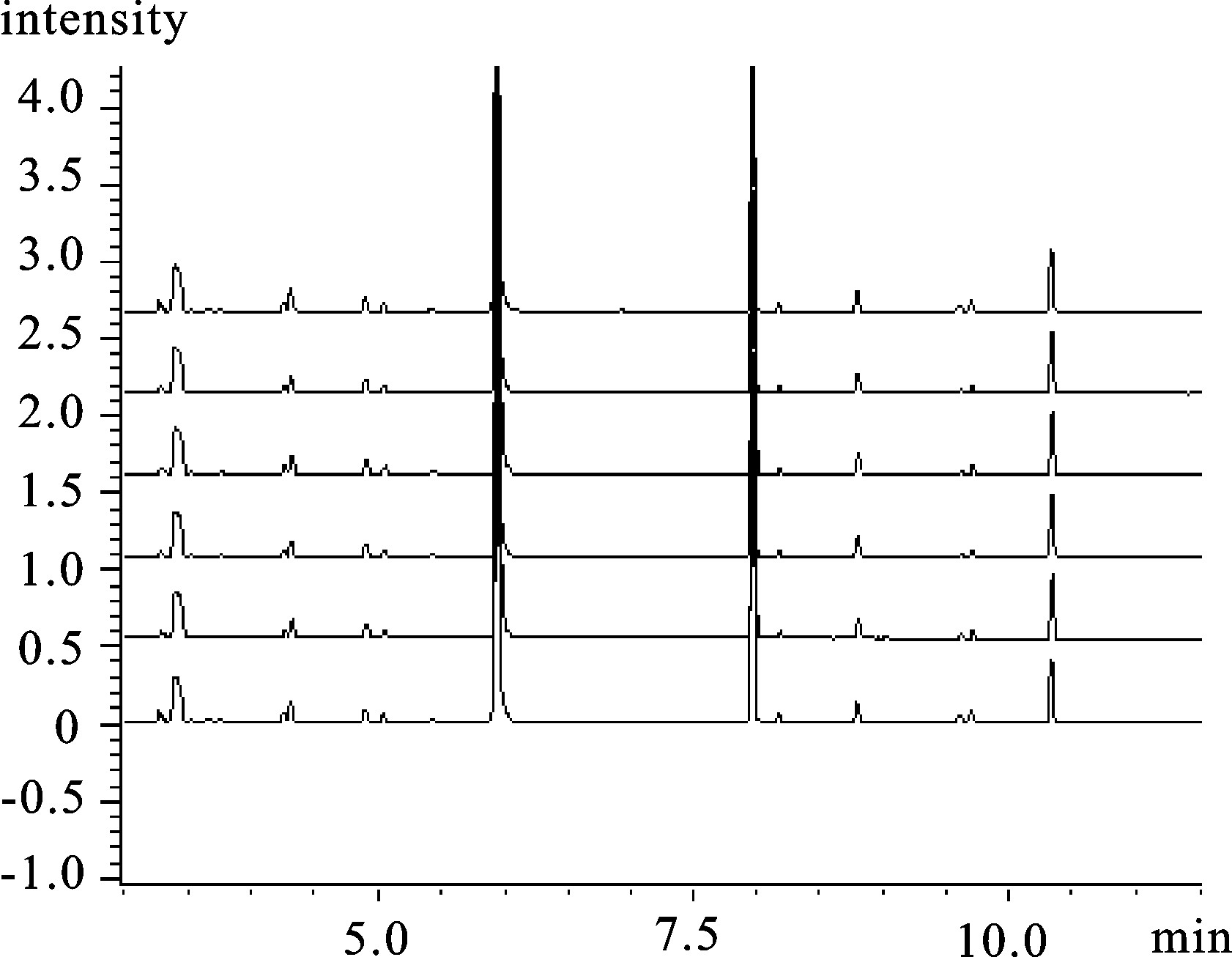
图 2 不同处理方法的亚硝胺峰响应强度
a. N-二甲基亚硝胺;b. N-二乙基亚硝胺;c. N-二丙基亚硝胺;d. N-亚硝基吡咯烷
Figure 2. Response intensity by different N-nitrosamines
a. N-dimethylnitrosamine; b. N-diethylnitrosamine; c. N-nitrosamines; d. N-nitroso-pyrrolidine
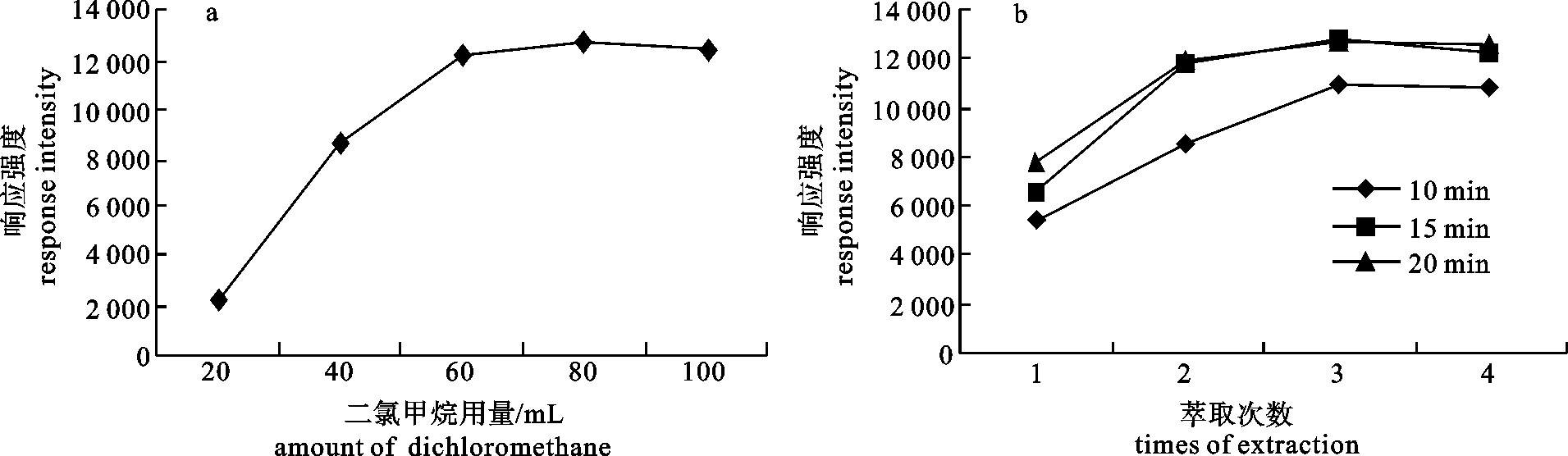
![]()
图 3 不同二氯甲烷用量(a)和不同萃取时间及次数(b)对萃取效果的影响
Figure 3. Effect of different methylene chlorides(a), different extraction time and times(b)on response intensity
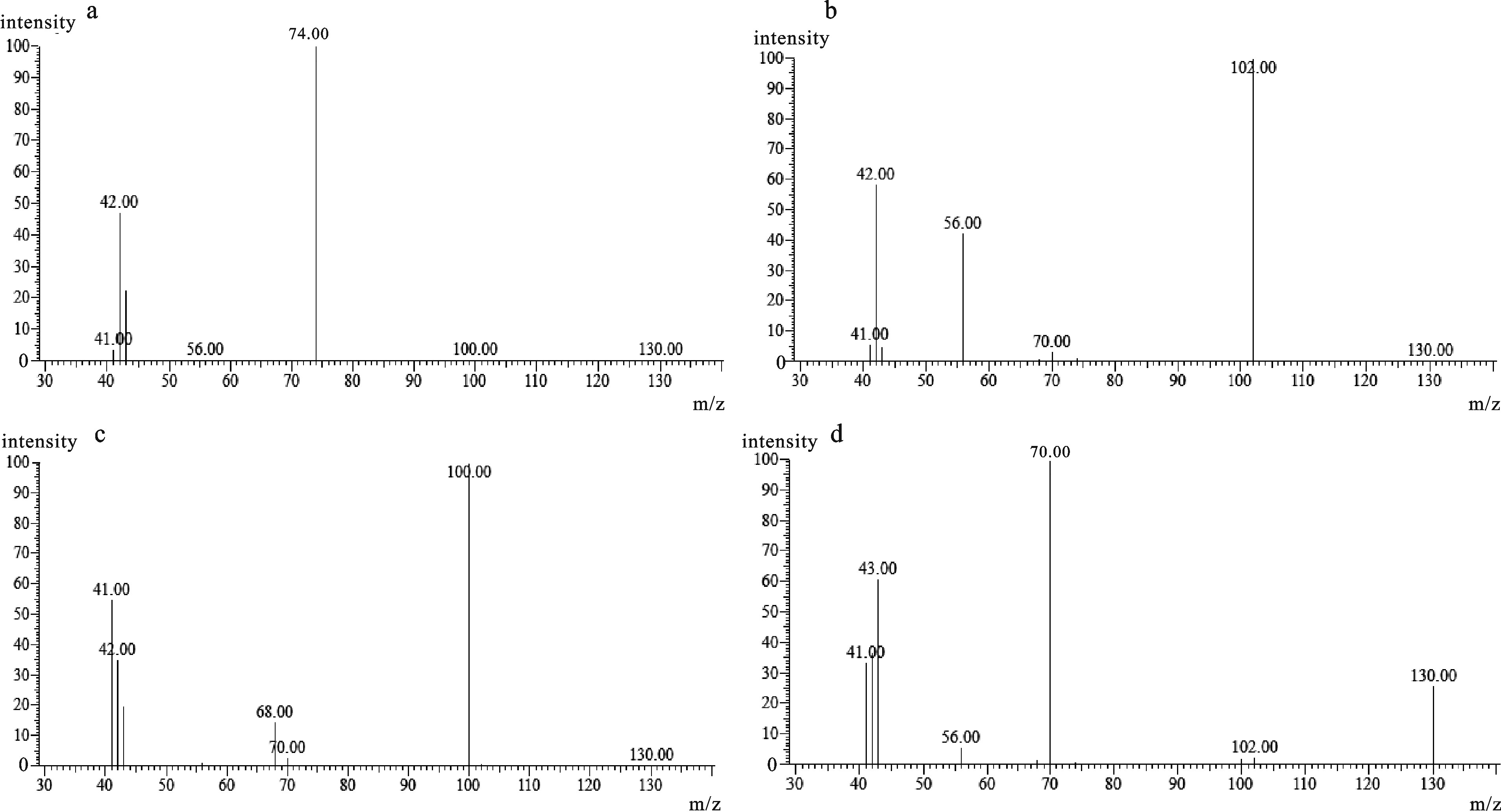
![]()
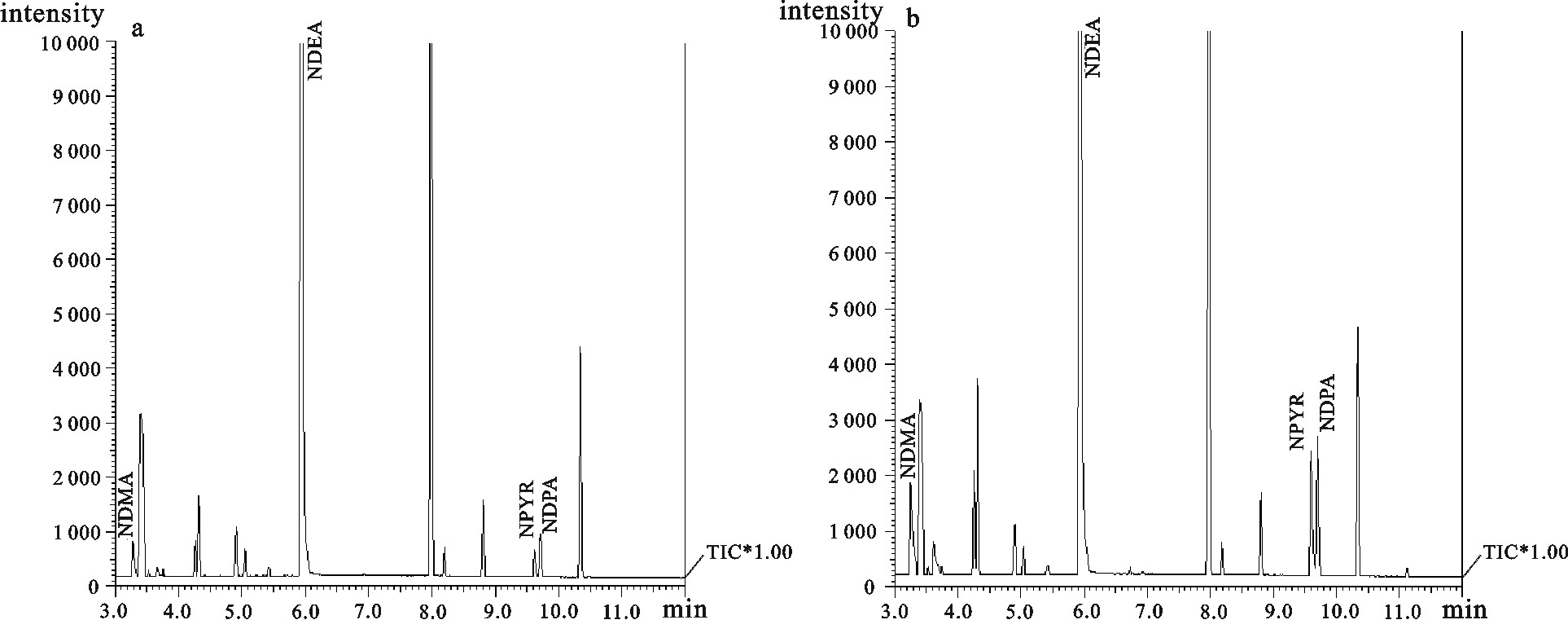
图 4 N-亚硝胺总离子图
a. N-二甲基亚硝胺;b. N-二乙基亚硝胺;c. N-二丙基亚硝胺;d. N-亚硝基吡咯烷
Figure 4. Total ion map of N-nitrosamines
a. N-dimethylnitrosamine; b. N-diethylnitrosamine; c. N-nitrosamines; d. N-nitroso-pyrrolidine
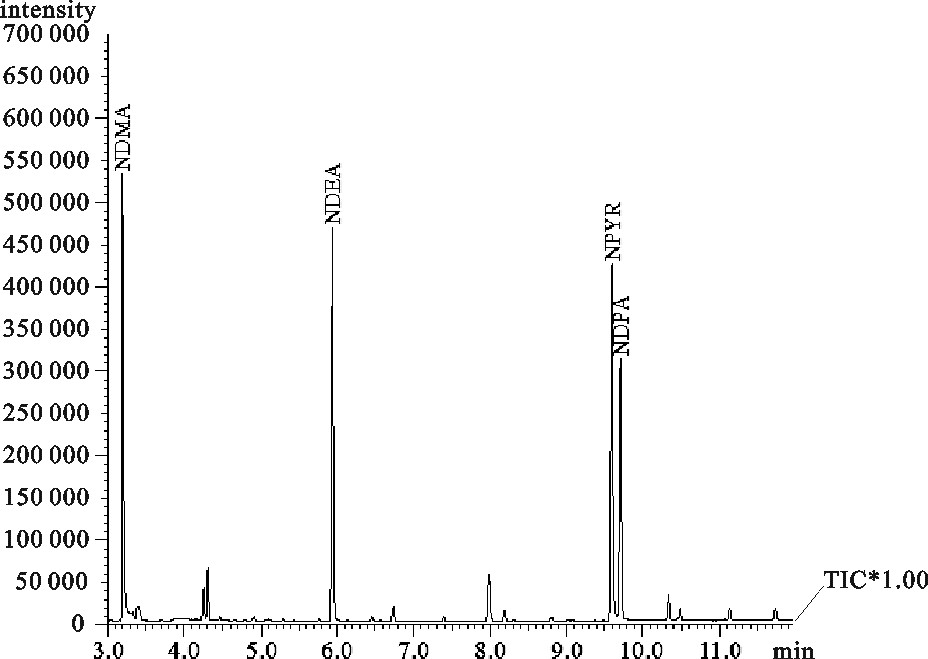
![]()
图 6 咸鱼样品添加4种混合N-亚硝胺标准品色谱图
Figure 6. Chromatogram of salted fish added 4 standard N-nitrosamines mixtures
表 1 4种挥发性N-亚硝胺的标准曲线及线性范围
Table 1 Standard curve and linear range of 4 volatile nitrosamines
N-亚硝胺
N-nitrosamine定量离子
m/z线性方程
linear regression equation相关系数(R)
correlation coefficient线性范围/μg·mL-1
linear rangeN-二甲基亚硝胺NDMA 74 y=3×10-5x+0.423 6 0.999 3 0~10 N-二乙基亚硝胺NDEA 102 y=4×10-5x+0.436 3 0.999 2 0~10 N-亚硝基吡咯烷NPYR 100 y=3×10-5x+0.460 2 0.999 1 0~10 N-二丙基亚硝胺NDPA 70 y=6×10-5x+0.368 5 0.999 2 0~10  下载: 导出CSV
下载: 导出CSV
表 2 各样品中N-亚硝胺质量分数(X +SD)
Table 2 Content of 4 N-nitrosamines
μg·kg-1 咸鱼样品名称
salted fishw(N-二甲基亚硝胺)
NDMAw(N-二乙基亚硝胺)
NDEAw(N-亚硝基吡咯烷)
NPYRw(N-二丙基亚硝胺)
NDPA总量
total大黄鱼(Pseudosciaena crocea) 2.11±0.40 0.130±0.14 未检出 4.17±0.51 6.42±0.15 三牙鱼(Otolithes ruber) 1.81±0.32 0.008±0 2.31±0.50 未检出 4.13±0.32 金仓鱼(Pomfret) 2.57±0.52 0.520±0.31 0.03±0.01 1.69±0.24 4.82±0.20 多春鱼(Capelin) 3.75±0.74 未检出 0.29±0.11 0.85±0.17 4.89±0.44
下载: 导出CSV
表 3 回收率试验(n=6)
Table 3 Recovery experiment
N-亚硝胺
N-nitrosamine加标量/μg·mL-1
adding standard回收率/%
recovery相对标准偏差/%
RSDN-二甲基亚硝胺NDMA 2.5 71.20 0.65 5.0 70.14 1.32 10.0 73.88 3.96 N-二乙基亚硝胺NDEA 2.5 73.11 1.12 5.0 75.65 2.74 10.0 80.10 1.04 N-亚硝基吡咯烷NPYR 2.5 75.22 2.15 5.0 78.72 1.26 10.0 76.41 0.98 N-二丙基亚硝胺NDPA 2.5 70.27 1.04 5.0 71.72 2.63 10.0 75.23 3.45
下载: 导出CSV
表 4 仪器和方法检出限
Table 4 Determination limit of instrument and method
N-亚硝胺
N-nitrosamine仪器检测限/ng·mL-1
detection limit of instrument方法检出限/μg·kg-1
detection limit of methodN-二甲基亚硝胺NDMA 0.772 0.038 6 N-二乙基亚硝胺NDEA 0.454 0.022 7 N-亚硝基吡咯烷NPYR 0.632 0.031 6 N-二丙基亚硝胺NDPA 0.956 0.047 8
下载: 导出CSV
-
[1] 何计国, 甄润英. 食品卫生学[M]. 北京: 中国农业大学出版社, 2003: 89-100. HE Jiguo, ZHEN Runying. Food hygiene[M]. Beijing: China Agricultural University Press, 2003: 89-100. (in Chinese)
[2] 胡丽芳, 尹德凤, 周瑶敏, 等. 气质联用法测定咸鱼中N-二甲基亚硝胺含量[J]. 江西农业学报, 2009, 21(9): 135-136. doi: 10.3969/j.issn.1001-8581.2009.09.044 HU Lifang, YIN Defeng, ZHOU Yaomin, et al. Determination of N-dimethylnitrosamine in salt fish by gas chromatography-mass spectrometry[J]. Acta Agriculturae Jiangxi, 2009, 21(9): 135-136. (in Chinese) doi: 10.3969/j.issn.1001-8581.2009.09.044
[3] 丁红梅, 陈彬, 杨兴龙, 等. 气质联用法测定生食水产品中挥发性N-亚硝胺[J]. 食品与机械, 2010, 26(6): 54-69. doi: 10.3969/j.issn.1003-5788.2010.06.016 DING Hongmei, CHEN Bin, YANG Xinglong, et al. Determination of volatile raw aquatic products of N-nitrosaminesby gas chromatography-mass pectrometry[J]. Food Machinery, 2010, 26(6): 54-69. (in Chinese) doi: 10.3969/j.issn.1003-5788.2010.06.016
[4] 马俪珍, 南庆贤, 方长法. N-亚硝胺类化合物与食品安全性[J]. 农产品加工学刊, 2005(12): 8-12. doi: 10.3969/j.issn.1671-9646-B.2005.12.002 MA Lizhen, NAN Qingxian, FANG Changfa. N-nitrosmine compounds and food safety[J]. Farm Prod Process, 2005(12): 8-12. (in Chinese) doi: 10.3969/j.issn.1671-9646-B.2005.12.002
[5] MITACEK E J, BRUNNEMANN K D, SUTTAJIT M, et al. Exposureto N-nitroso compounds in a population of high liver cancer regions in Thailand: volatilenitrosa-mine (VNA) levels in Thaifood[J]. Food Chem Toxicol, 1999, 37(4): 297-305. doi: 10.1016/S0278-6915(99)00017-4
[6] 蔡一新, 林升清, 林生金. 福建省部分食品中N-亚硝胺含量调查结果分析[J]. 中国卫生检验杂志, 1997, 7(6): 356-358. https://kns.cnki.net/kcms2/article/abstract?v=MdENDFpkZq4mw_wziPZc4tmbuCvY97oWlxoY9Y0_nKBSq8g3DSW6GKZ--hrKnJbklzW1OeaBBXuGfnxiBBOvaUtWrY_HFcVQ62E1ZOLuSnJgBcd1mKdxzM5UGk93fZbpPKufFwp4YZv2KqDIAdmTXwvC4J4q9Er7YUCSKQw1du6c8gkZlbNbg1gCs4l42Uie&uniplatform=NZKPT&language=CHS CAI Yixin, LIN Shengqing, LIN Shengjin. N-nitrosamine content analysis of the survey in part of the food from Fujian province[J]. Chin J Health Lab Technol, 1997, 7(6): 356-358. (in Chinese) https://kns.cnki.net/kcms2/article/abstract?v=MdENDFpkZq4mw_wziPZc4tmbuCvY97oWlxoY9Y0_nKBSq8g3DSW6GKZ--hrKnJbklzW1OeaBBXuGfnxiBBOvaUtWrY_HFcVQ62E1ZOLuSnJgBcd1mKdxzM5UGk93fZbpPKufFwp4YZv2KqDIAdmTXwvC4J4q9Er7YUCSKQw1du6c8gkZlbNbg1gCs4l42Uie&uniplatform=NZKPT&language=CHS
[7] 樊丽琴. 咸鱼腌制过程中N-亚硝胺及其前体物质的变化规律研究[D]. 湛江: 广东海洋大学, 2009: 16-17. 10.7666/d.y1552051 FAN Liqin. Study on the changing regularity of N-nitrosamine and its precursor substance in pickling salted fish[D]. Zhanjiang: Guangdong Ocean University, 2009: 16-17. (in Chinese) 10.7666/d.y1552051
[8] 刘法佳, 吴燕燕, 李来好, 等. 降低腌制食品中亚硝酸盐含量的研究进展[J]. 广东农业科学, 2011, 38(1): 165-167. doi: 10.3969/j.issn.1004-874X.2011.01.061 LIU Fajia, WU Yanyan, LI Laihao, et al. Development of reducing of nitrite in salted food[J]. Guangdong Agric Sci, 2011, 38 (1): 165-167. (in Chinese) doi: 10.3969/j.issn.1004-874X.2011.01.061
[9] 吴燕燕, 刘法佳, 李来好, 等. 改良离子色谱法测定咸鱼中亚硝酸盐的研究[J]. 南方水产科学, 2011, 7(6): 1-6. doi: 10.3969/j.issn.2095-0780.2011.06.001 WU Yanyan, LIU Fajia, LI Laihao, et al. Determination of nitrite in salted fishes by the improved ion chromatography[J]. South China Fish Sci, 2011, 7(6): 1-6. (in Chinese) doi: 10.3969/j.issn.2095-0780.2011.06.001
[10] 孙效晖, 韩一鸣, 林弟雄, 等. GB 10138-2005盐渍鱼卫生标准[S]. 北京: 中国标准出版社, 2005. SUN Xiaohui, HAN Yiming, LIN Dixiong, et al. GB 10138-2005. Hygienic standard for salted fish[S]. Beijing: Standards Press of China, 2005. (in Chinese)
[11] 魏法山, 徐幸莲, 周光宏. 挥发性N-亚硝基化合物的分析方法[J]. 食品科学, 2008, 29(7): 479-483. doi: 10.3321/j.issn:1002-6630.2008.07.109 WEI Fashan, XU Xinglian, ZHOU Guanghong. Determination method of volatile N-nioso compounds[J]. Food Sci, 2008, 29(7): 479-483. (in Chinese) doi: 10.3321/j.issn:1002-6630.2008.07.109
[12] 王瑞, 马俪珍, 方长发. 毛细管气相色谱法对冷却猪肉中挥发性N-亚硝胺类化合物含量的测定分析[J]. 天津农学院学报, 2006, 13(1): 10-13. doi: 10.3969/j.issn.1008-5394.2006.01.003 WANG Rui, MA Lizhen, FANG Changfa. Application of gas chromatography by capillary tube to determine volatilizable N-nitrosamine compound in cooled pork[J]. J Tianjin Agric Univ, 2006, 13(1): 10-13. (in Chinese) doi: 10.3969/j.issn.1008-5394.2006.01.003
[13] 张秋菊, 郭祖鹏, 李明珠, 等. 顶空固相微萃取-气相色谱-质谱法测定7种亚硝胺类化合物[J]. 中国卫生检验杂志, 2009, 19(6): 1234-1236. https://kns.cnki.net/kcms2/article/abstract?v=MdENDFpkZq4nArXXU3X4WZc8ylJDUb7tavi5rIpoDQQadAKBvKGcvCNgCMlT5mMG03FD4NyoQyFfCZ9w76HiyMZJg6cwsS5-VtJg1Rs1xG80md1B8DrUGrSp8YeIBpxbrmK_8wpQPWjkMQdJQPpUX4FAOyq5Nt1Y1MbrEjd8j68CG15mtG8yWmnMrz3shNSf&uniplatform=NZKPT&language=CHS ZHANG Qiuju, GUO Zupeng, LI Ming zhu, et al. Determination of seven N-nitrosamine compounds by HS-SPME-GC-MS[J]. Chin J Health Lab Technol, 2009, 19(6): 1234-1236. (in Chinese) https://kns.cnki.net/kcms2/article/abstract?v=MdENDFpkZq4nArXXU3X4WZc8ylJDUb7tavi5rIpoDQQadAKBvKGcvCNgCMlT5mMG03FD4NyoQyFfCZ9w76HiyMZJg6cwsS5-VtJg1Rs1xG80md1B8DrUGrSp8YeIBpxbrmK_8wpQPWjkMQdJQPpUX4FAOyq5Nt1Y1MbrEjd8j68CG15mtG8yWmnMrz3shNSf&uniplatform=NZKPT&language=CHS
[14] 方长发, 马俪珍, 刘会平, 等. 固相微萃取技术及其在N-亚硝胺分析中的应用[J]. 肉类研究, 2008(4): 49-50. doi: 10.3969/j.issn.1001-8123.2008.04.014 FANG Changfa, MA Lizhen, LIU Huiping, et al. Solid phase microextraction (SPME) and its application in nitrosamine analysis[J]. Meat Res, 2008(4): 49-50. (in Chinese) doi: 10.3969/j.issn.1001-8123.2008.04.014
[15] 陶燕飞, 黄红林, 张桃芝. 啤酒中N-亚硝胺的SPME-GC-MS分析[J]. 分析测试学报, 2003, 22(5): 82-84. doi: 10.3969/j.issn.1004-4957.2003.05.024 TAO Yanfei, HUANG Honglin, ZHANG Taozhi. Determination of volatile N-nitrosocompounds in beer by SPME-GC-MS[J]. J Instrumental Anal, 2003, 22(5): 82-84. (in Chinese) doi: 10.3969/j.issn.1004-4957.2003.05.024
-
期刊类型引用(5)
1. 范嗣刚,黄皓,王鹏飞,闫路路,赵超,张博,邱丽华. 基于cox1序列的中国6个花鲈野生群体遗传多样性. 广东海洋大学学报. 2022(03): 11-17 .  百度学术
百度学术
2. 胡玉婷,江河,段国庆,凌俊,潘庭双,周华兴,汪焕. 基于线粒体标记的滁州鲫遗传多样性和遗传结构. 安徽农学通报. 2021(24): 12-14+61 . 百度学术
3. 董传举,李学军,孙效文. 我国鲫种群遗传多样性及起源进化研究进展. 水产学报. 2020(06): 1046-1062 . 百度学术
4. 彭敏,王大鹏,施军,韩耀全,雷建军,李育森,吴伟军,何安尤. 西江流域卷口鱼线粒体D-loop序列的遗传多样性分析. 渔业科学进展. 2020(05): 30-37 . 百度学术
5. 颜岳辉,丁雪梅,李强,李旭,唐利洲. 珠江源头入侵种波氏吻虾虎的遗传多样性分析. 四川动物. 2019(03): 263-270 . 百度学术
其他类型引用(3)





计量
- 文章访问数: 4095
- HTML全文浏览量: 185
- PDF下载量: 3691
- 被引次数: 8
 粤公网安备 44010502001741号
粤公网安备 44010502001741号
