
Study on determination of Tilmicosin residue in aquatic products
-
摘要:
文章探讨了用高效液相色谱(HPLC)测定水产品中替米考星残留量的方法。通过对分析条件的优化,确定了最佳检测参数,并从提取和净化2个方面确立了前处理的最佳步骤与条件。通过验证不同基质试样的加标回收试验,结果显示,在20~200 μg · kg-1的添加水平下回收率为77.22%~93.25%,相对标准偏差(RSD)小于5%,定量检出限(LOQ)为20 μg · kg-1。分析结果表明,所建立的方法灵敏度高、准确性好且应用范围广,可用于日常水产品中替米考星残留量的检测分析。
Abstract:A HPLC method for determining Tilmicosin in aquatic products was discussed in this paper. The optimal detection parameters were determined by optimizing the analytical conditions; the best procedure and factors of pretreatment were determined in the aspects of extraction and clean-up. Through the experimental verification of different sample substrates, the recovery of standard addition reached 77.22%~93.25% and RSD was less than 5% with the addition of 20~200 μg · kg-1. Moreover, the limit of quantitation was 20 μg · kg-1. The results showed that this method had advantages of high test sensitivity, accuracy and wide range of application. It could be used for routine detection and analysis of Tilmicosin residue in aquatic products.
-
Keywords:
- aquatic products /
- Tilmicosin /
- residue /
- HPLC
-
pH调节法提取蛋白质早期应用于鱼白肉和碎鱼肉中提取、分离鱼蛋白。20世纪末,美国学者就报道了一种提取动物蛋白的新方法,即pH调节法或称酸碱法、等电点溶解/沉淀法[1]。该法是根据蛋白质在不同酸碱度中溶解度的异同,利用蛋白质在等电点附近溶解度最低的原理将蛋白质沉淀并回收。整个提取过程具有操作温度低、蛋白质变性程度小、功能特性保持较完全等优点。此后,采用等电点法提取分离动物蛋白的报道逐渐增多。UNDELAND等[2]报道了利用等电点法从鲱(Clupea harengus)鱼肉中提取的分离蛋白具有更好的色泽和优良的凝胶强度。CHOI和PARK[3]对酸处理(pH 5.0)回收太平洋鳕(Gadus macrocephalus)蛋白的研究表明,与传统水洗法相比,酸处理法回收蛋白的得率高、组织蛋白酶的活性强,但部分蛋白,如肌球蛋白和肌动蛋白在提取过程中发生了降解。等电点法主要应用于以鱼白肉、碎鱼肉和鱼下脚料为原料的鱼蛋白提取[4-7]。GIGLIOTTI等[8]采用了等电点法从南极磷虾(Euphaususa superba)中分离提取蛋白,并对其营养价值和安全性进行了分析。其蛋白质含量为78%(干基),安全性与酪蛋白无明显差异。
在甲壳动物中,绝大多数类胡萝卜素都以类胡萝卜蛋白(类胡萝卜素结合高密度脂蛋白)的形式存在[9]。该蛋白被认为有2种作用,改善色素的水溶性和提供颜色。甲壳动物的类胡萝卜素蛋白分为3种[10-11]:1)类胡萝卜脂蛋白。主要分布在甲壳动物的卵和卵巢中,但在血液和表皮中也有少量存在。它们使组织呈现蓝色、绿色和紫色;2)从甲壳动物外壳中发现的几丁质类胡萝卜素。它们由几丁质和类胡萝卜素通过Schiff碱键结合而成或是由几丁质的氮端和类胡萝卜素上的酮环结合而成;3)类胡萝卜结合蛋白。主要是虾青素结合脱辅基蛋白而生成的虾青蛋白,它们主要存在于甲壳动物的体表结构中,为其提供保护色。
虾壳在虾熟制过程中会发生红变现象,这是由于虾青蛋白受热变性,释放了与其共价结合的类胡萝卜素而引起的[12]。虾壳的色泽不仅会左右人们的购买,而且也在一定程度上反映了整虾的新鲜程度[13]。传统方法提取虾青蛋白主要是经硫酸铵分级沉淀后,采用高效液相色谱进一步纯化[14]。不仅试验试剂消耗大、目标蛋白得率低,并且温度对目标蛋白的提取影响大。该试验采用pH调节法对比传统硫铵盐析法从凡纳滨对虾壳中提取β虾青蛋白,并对所得蛋白在回收率、纯度和光谱性质等方面进行比较,旨在对pH调节法提取虾青蛋白的可行性进行分析讨论,为有效提取、利用甲壳动物甲壳中的虾青蛋白提供参考。
1. 材料与方法
1.1 材料
凡纳滨对虾(Litopenaeus vannamei)虾壳(亚洲水产,湛江产),运回实验室后立即用冰水洗净、4 ℃下干燥、粉碎、称质量。
1.2 仪器与试剂
UV-1800紫外可见分光光度计(日本岛津出品),CR22GⅡ型高速冷冻离心机(日本日立出品),LC-20AD高效液相色谱仪(日本岛津出品),AE-7300型电泳仪(日本帝国理化出品),Jasco-725型圆二色光谱仪(日本Jasco出品),Alpha 2-4型真空冷冻干燥机(德国Martin Christ出品),Ettan IPGphorⅡ型等电聚焦仪(美国GE Healthcare出品),F-52型pH计(日本崛场出品),Immobiline DryStrip pH 3-10 NL、13 cm型胶条(美国GE Healthcare出品)。试验用水为超纯水,磷酸氢二钠、磷酸二氢钠、EDTA和CHAPS等试剂均为国产分析纯。
1.3 方法
1.3.1 盐析法提取β虾青蛋白
参考ZAGALSKY[14]。将虾壳用预冷却的质量分数为10%的EDTA溶液浸泡[m(虾壳,g):V(EDTA, mL)=25:1 000]10 h(4 ℃)。过滤后向滤液中加入55%饱和度的硫酸铵,4 ℃静置10 h。离心(10 000 g,4 ℃,20 min)后,用100 mL、50 mmol·L-1磷酸盐缓冲液(pH 7.0)溶解沉淀并加入20 %饱和度的硫酸铵,4 ℃静置10 h。二次离心后弃沉淀并向清液中加入50%饱和度的硫酸铵,4 ℃静置10 h后离心所得沉淀即为粗蛋白。将多次提取的粗蛋白收集后,通过真空冷冻干燥浓缩、保存。
在使用高效液相色谱纯化前,需用保存液(50 mmol·L-1,pH 7.0磷酸盐缓冲液)透析数次,以尽量减少硫酸铵对纯化的影响。试验选用的是离子交换柱(5 mm×50 mm Mono Q阴离子交换柱)。流动相:溶剂A为50 mmol·L-1磷酸盐缓冲液(pH 7.0),溶剂B为含1.0 mol·L-1氯化钠的50 mmol·L-1磷酸盐缓冲液(pH 7.0)。梯度洗脱程序为10~80 min内溶剂B的体积从0上升至70%,90 min时增加至100%并维持10 min,115 min时降为0;流速为0.5 mL·min-1,进样量1 mL。
1.3.2 pH调节法提取β虾青蛋白
将预处理后的凡纳滨对虾虾壳用预冷却的质量分数为10%的EDTA溶液浸泡10 h、过滤。提取液pH设置为3.0~11.0,以1.0为pH变化梯度。将滤液分为2份,分别用2 mol·L-1的盐酸(HCl)调节至3.0(酸处理)和2 mol·L-1的氢氧化钠(NaOH)调节pH至11.0(碱处理)。4 ℃、10 000 g离心17 min,得到不同pH下的溶出物和未溶物。
用2 mol·L-1的HCl或NaOH将溶出物的pH调节至等电点后4 ℃、10 000 g离心17 min,得到目标蛋白和未回收的溶出物。
蛋白质溶解度和回收率计算公式为[15]:
$$ 蛋白质溶解度 (\%)=\frac{A}{C} \times 100 $$ $$ 蛋白质回收率 (\%)=\frac{A-A_1}{C} \times 100 $$ 其中A为不同pH处理后离心所得清液中虾青蛋白质量(mg)=清液中蛋白质质量浓度(mg·mL-1)×清液体积(mL);A1为等电点沉淀后,离心所得清液中的虾青蛋白质量(mg);C为总虾青蛋白质量(mg),C=EDTA脱钙后滤液中虾青蛋白质量浓度(mg·mL-1)×滤液体积(mL)。每组试验重复3次取平均值。
1.3.3 β虾青蛋白的基本性质分析
1) 等电聚焦电泳。将经盐析、离子交换色谱纯化后的蛋白质溶于50 mmol·L-1磷酸盐缓冲液中(pH 7.0),透析数次,以消除盐对试验结果的影响。向洁净、干燥的胶条槽(13 cm)内加入含1.0 mL β虾青蛋白纯品(170.14 mg)的再水化液250 mL,将撕去保护膜的IPG胶条置于胶条槽内(酸性端位于正极),尽量避免气泡的形成。最后加入IPG覆盖液以防止蛋白质样品的蒸发和尿素结晶的形成。等电聚焦电泳参数设定为500 V,60 min;1 000 V梯度,60 min;8 000 V梯度,150 min;8 000 V,25 min。电泳完成后经固定、染色、脱色后拍照、保存。2)SDS-PAGE电泳。预制胶浓度为5%~20%(Atto,日本产),Marker范围为10~250 kDa(Bio-Rad,美国产)。电泳完成后经考马斯亮蓝R250染色,脱色。3)凝胶过滤层析。对经过2种方法纯化后的蛋白质选用HiPrep 16/60 Sephacryl S-100 HR和S-300 HR预装柱(GE Healthcare,美国产)进一步确认其分子量。洗脱液为50 mmol·L-1、pH 7.0的磷酸盐缓冲液,流速为15 mL·h-1,标准品分子量为1.35~670 kDa(Bio-Rad,美国产)。4)蛋白质纯度测定。利用凯氏定氮法(GB/T 5009.5-2003)进行测定。每组试验重复3次取平均值。5)最大吸收波长的测定。利用紫外可见光谱仪对蛋白质的最大吸收波长进行测定。6)β虾青蛋白二级结构的测定。在Jasco-725型圆二色光谱仪上进行圆二色光谱测量,光源系统用氮气保护(流量为15 L·min-1),试验均采用2 mm光程石英样品池。测量参数:扫描波长范围200~240 nm,扫描时间50 nm·min-1,分辨率0.1 nm,响应时间1 s,累积次数3次,在室温下进行测定[16]。CD谱用平均残基摩尔椭圆度表示,单位deg·cm2·dmol-1。利用圆二色光谱仪附带的杨氏法二级结构分析软件对二级结构含量进行计算[17]。
2. 结果与分析
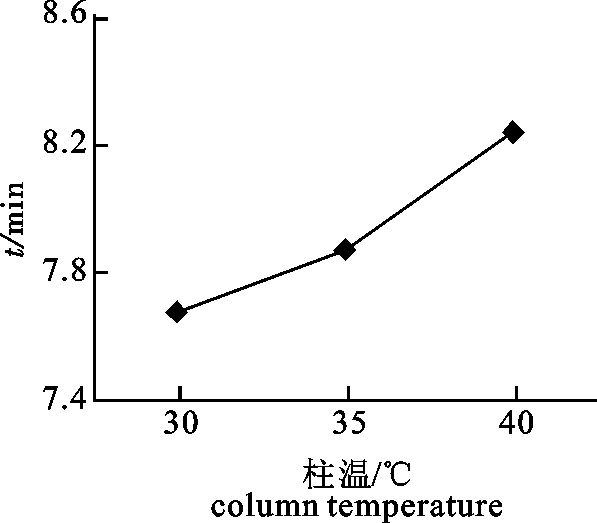
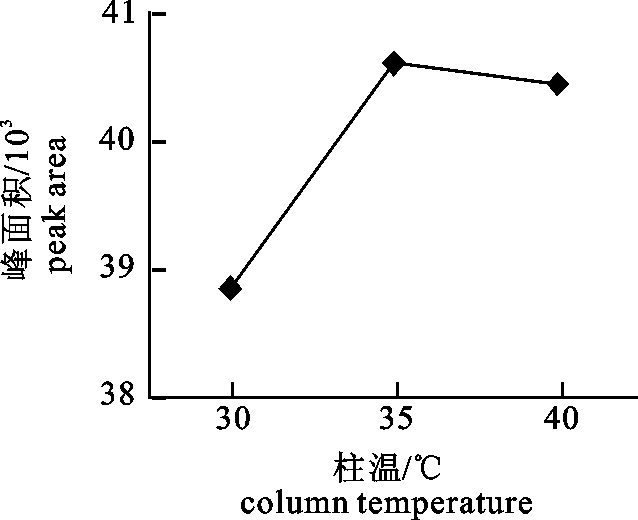
2.1 pH调节法分离蛋白质的回收率、溶解度和纯度
pH处理对蛋白质溶解度的影响呈现先下降后上升的趋势(图 1)。当pH为3.0~5.0时溶解度呈逐渐下降趋势;pH为5.0~11.0时溶解度呈上升趋势;5.0~9.0时上升较为缓慢。酸性溶液中蛋白质溶解度在pH 3.0时最大为60.5%,碱性溶液中蛋白质溶解度在pH 11.0时最大为55.7%;pH为5.0时溶解度最小为15.4%。
![]() 图 1 β虾青蛋白溶解度、回收率随pH的变化曲线Figure 1. Solubility and recovery rate of β crustacyanin at pH 3~11
图 1 β虾青蛋白溶解度、回收率随pH的变化曲线Figure 1. Solubility and recovery rate of β crustacyanin at pH 3~11pH调节法对蛋白质回收率的影响趋势与溶解度一致。最大回收率为pH 3.0时的47.5%,高于传统硫铵盐析的24.3%。同时,pH调节法所得β虾青蛋白的纯度经凯氏定氮法测得为78.23%,也高于盐析法的74.06%。
2.2 β虾青蛋白的光谱性质
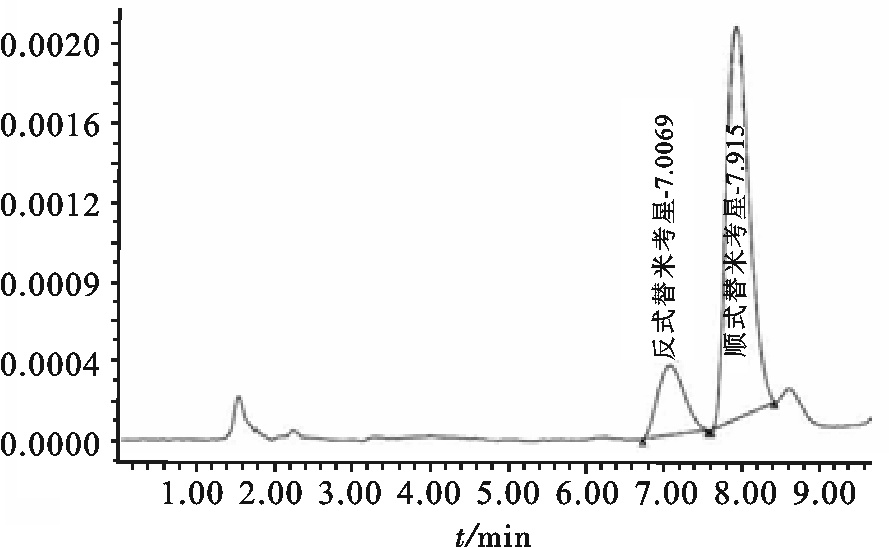
盐析法所得虾青蛋白粗品经高效液相分离纯化,按洗脱先后顺序共有3个峰:1)在20% B液条件下洗脱出,在520 nm处有最大吸收,被定义为α虾青蛋白;2)在40% B液条件洗脱出,在580 nm处有最大吸收,被定义为β虾青蛋白;3)在100% B液条件下被洗脱出,在500 nm处有最大吸收,被定义为虾青蛋白的变性产物(图 2)。这与ZAGALSKY [14]的研究一致,α虾青蛋白在0.25 mol·L-1浓度下被洗脱。而TIMME等 [18]对南非龙虾(Jasus lalandii)的研究认为α与β虾青蛋白的洗脱浓度分别为0.4 mol·L-1和0.2 mol·L-1 B液浓度,且最大吸收峰分别为525 nm和560 nm。产生洗脱差异的原因仍需进一步研究,初步推断是组成蛋白质的氨基酸序列和数量的不同所引起。
![]() 图 2 凡纳滨对虾壳中提取虾青蛋白的高效液相纯化色谱图Figure 2. Elution profile of crustacyanin from white leg shrimp shell by anion-exchange chromatography on a Mono Q 5/50GL column
图 2 凡纳滨对虾壳中提取虾青蛋白的高效液相纯化色谱图Figure 2. Elution profile of crustacyanin from white leg shrimp shell by anion-exchange chromatography on a Mono Q 5/50GL column图 3为不同提取方法获得的β虾青蛋白的紫外可见光谱图。可见2种方法获得的目标蛋白在最大吸收波长上无差异,均为580 nm。TIMME等[18]发现南非龙虾壳中提取的β虾青蛋白的最大吸收波长为560 nm;ZAGALSKY[14]在对无脊椎动物中提取的类胡萝卜蛋白研究后指出,美国龙虾壳中提取的β虾青蛋白的最大吸收波长为580~590 nm;CHAYEN等[19]研究发现欧洲龙虾(Homarus gammarus)壳中提取的β虾青蛋白的最大吸收波长为585 nm。不同地区虾壳中提取的β虾青蛋白最大吸收波长差异的原因仍需进一步研究。
![]() 图 3 2种方法提取所得β虾青蛋白紫外可见光谱图Figure 3. Visible absorption spectra of β crustacyanin extracted by pH-shifting and ammonium sulfate precipitation
图 3 2种方法提取所得β虾青蛋白紫外可见光谱图Figure 3. Visible absorption spectra of β crustacyanin extracted by pH-shifting and ammonium sulfate precipitation标准蛋白质二级结构的圆二色光谱为:α螺旋在222 nm和208 nm处呈现负峰,在192 nm附近显示一个正峰;β折叠在215 nm处呈负峰,在198 nm附近有一正峰;无规则卷曲则在198 nm有一负峰,在220 nm附近有一正峰。试验得到的圆二色光谱可以看作一定百分比的α螺旋、β折叠和无规则卷曲的线性迭加图谱[20]。通过对比标准二级结构的远紫外圆二色光谱图,试验结果显示β虾青蛋白二级结构主要是α螺旋(盐析法),含量为65%~70% (25 ℃)(图 4)。pH调节法所得β虾青蛋白与传统盐析法提取的β虾青蛋白在二级结构含量上无明显差异,分别为70.1%和67%。
![]() 图 4 pH调节法和硫铵盐析法所得β虾青蛋白的远紫外圆二色光谱图Figure 4. Far-UV CD spectra of β crustacyanin extracted by pH-shifting and ammonium sulfate precipitation
图 4 pH调节法和硫铵盐析法所得β虾青蛋白的远紫外圆二色光谱图Figure 4. Far-UV CD spectra of β crustacyanin extracted by pH-shifting and ammonium sulfate precipitation2.3 β虾青蛋白的等电点与分子量
盐析法所得β虾青蛋白等电聚焦电泳仅显示1条条带(图 5),证明凯氏定氮中盐析法所得蛋白质纯度为74.06%是可靠的。以标准蛋白质等电点为对照计算得知β虾青蛋白等点电为5.6,这也符合pH 5.0~6.0时蛋白质溶解度最低。
![]() 图 5 盐析法提取的β虾青蛋白纯品等电聚焦电泳图(蛋白质质量浓度为0.12 mg·mL-1)Figure 5. Isoelectric focusing electrophoresis of β crustacyanin with concentration of 0.12 mg·mL-1 extracted via ammonium sulfate precipitation
图 5 盐析法提取的β虾青蛋白纯品等电聚焦电泳图(蛋白质质量浓度为0.12 mg·mL-1)Figure 5. Isoelectric focusing electrophoresis of β crustacyanin with concentration of 0.12 mg·mL-1 extracted via ammonium sulfate precipitation凝胶色谱结果显示,在20% B液下收集的520 nm处有最大吸收峰,被定义为α虾青蛋白的物质,分子量为380 kDa;在40% B液下收集的580 nm处有最大吸收峰,被定义为β虾青蛋白的物质,分子量为45 kDa,这与TIMME等[18]的研究结果较为一致。
聚丙烯酰胺凝胶电泳粗蛋白显示3个特征条带区(图 6),分布在相对分子量为350 kDa、75 kDa和37 kDa处。β虾青蛋白是由2个分子量约为20 kDa的亚基组成的二聚物[14, 19],因此推断37 kDa附近的条带为β虾青蛋白。而α虾青蛋白是由8个β型组合而成的一个八面体结构[14, 19],因此350 kDa的条带推断为α虾青蛋白。75 kDa的条带仍需进一步研究,推测为α型的降解产物[19]或是纯化过程中得到的其他蛋白质亚基[14]。2种方法纯化后的蛋白质在37 kDa附近的条带均被稀释,说明SDS-PAGE不适合用来鉴定β虾青蛋白分子量。
![]() 图 6 虾青蛋白聚丙烯酰胺凝胶电泳图泳道从左至右为Marker、粗虾青蛋白、盐析法所得β虾青蛋白、pH调节法所得β虾青蛋白Figure 6. Gel electrophoresis of polyacrylamide from crustacyaninLanes from left to right indicate Marker, crude crustacyanin, β crustacyanin extracted by ammonium sulfate precipitation and β crustacyanin extracted by pH-shifting
图 6 虾青蛋白聚丙烯酰胺凝胶电泳图泳道从左至右为Marker、粗虾青蛋白、盐析法所得β虾青蛋白、pH调节法所得β虾青蛋白Figure 6. Gel electrophoresis of polyacrylamide from crustacyaninLanes from left to right indicate Marker, crude crustacyanin, β crustacyanin extracted by ammonium sulfate precipitation and β crustacyanin extracted by pH-shifting3. 讨论
蛋白质的溶解度对优化酸碱法回收蛋白质有决定性作用。在酸性或碱性条件时,蛋白质由于携带不同电荷会发生沉淀或溶解的现象[21]。极端pH条件下,由于强烈的排斥作用蛋白质很难发生沉淀(尽管此时已有蛋白质发生变性),它会与水分子结合而使溶解度升高。当溶液pH处于蛋白质等电点附近时,蛋白质净电荷为零而产生聚合现象使溶解度降低[19]。溶解度高有利于将目标蛋白从杂质中分离出来,而等电点时的低溶解度则有利于提高目标蛋白的回收率[22]。从凡纳滨对虾壳中提取的β虾青蛋白溶解度曲线呈U型,这与尼罗罗非鱼(Oreochromis niloticus)[23]和南极磷虾[15]肌肉中回收的蛋白质溶解度曲线一致。随着pH的降低,溶解度逐渐增加的原因是蛋白质在酸度系数2.5~7.0时比7.0~11.0含有更多的离子化基团[2]。
β虾青蛋白通过萃取和沉淀从凡纳滨对虾壳中获得,在pH 3.0和11.0时其回收率约为40%~47%。PALAFOX等[22]使用pH调节法从巨型鱿鱼(Dosidicus gigas)中提取肌肉蛋白的回收率约为75%;KRISTINSSON等[24]采用pH调节法从石首鱼(Micropogonias undulates)中提取肌肉蛋白的回收率约为65%~78%。β虾青蛋白回收率较低的原因是虾壳主要由虾青素和几丁质构成,蛋白质含量相对较低[25]; CHEN和JACZYNSKI[26]报道了pH调节法提取蛋白质的回收率与溶液离子强度有关。类胡萝卜素在结合载脂蛋白后会增强其水溶性[10, 12],同时蛋白质在pH 3.0和11.0时的高溶解度会进一步提高β虾青蛋白的回收率。
pH调节法在目标蛋白的纯度和产量上均优于传统盐析法。这主要是由于pH调节法可以减少提取步骤从而降低杂蛋白的产量和不利影响。同时,蛋白质具有酸碱基团的特性也有利于增加产量、提高目标蛋白纯度。
虾青蛋白对虾壳熟制过程中的颜色变化发挥着重要作用,因此提高蛋白质的提取率对深入了解和利用虾青蛋白具有重要意义。pH调节法提取β虾青蛋白在蛋白质回收率、纯度上均优于传统盐析法,蛋白质分子量、二级结构也无明显差异。虽然等电点的确认需要通过传统盐析法得到纯品,而且在等电点法提取时酸碱会在一定程度上引起蛋白质变性,但该法简单、快捷,蛋白质产量和纯度均优于传统方法, 同时试剂消耗少,试验条件要求低。因此,pH调节法可以作为从虾壳中提取β虾青蛋白的参考方法。
-
表 1 梯度洗脱程序
Table 1 Procedure of gradient elution
t/min A/% B/% 0 25 75 3 25 75 9 35 65 10 25 75 15 25 75  下载: 导出CSV
下载: 导出CSV
表 2 不同提取剂比较结果
Table 2 Results of different extract solvents
提取剂
extract solvent回收率/%
recovery备注
remark乙酸乙酯
ethylacetate55~97 对于脂肪含量较低的虾等样品,乙酸乙酯的提取效果最好;但对于鳗鱼等高脂肪样品,由于油脂不易除去,对回收率影响大 乙腈
acetonitrile75~96 提取效果好,油脂易去除 酸化乙腈
aidified cetonitrile70~87 利于除蛋白,但酸性条件下目标物稳定性较差 甲醇
methanol73~89 提取效果一般,油脂易去除 偏磷酸-甲醇
metaphosphoric acid-methanol71~94 偏磷酸不易溶于水,且溶于水后生成的正磷酸有剧毒,危害大
下载: 导出CSV
表 3 回收率试验结果
Table 3 Results of recovery
添加水平/μg·kg-1
added concentration回收率/%
recovery平均回收率/%
average recovery标准偏差
SD相对标准偏差/%
RSD20 78.68 79.69 3.10 3.9 77.22 83.17 100 87.32 91.06 3.26 3.6 92.61 93.25 200 84.62 85.94 1.81 2.1 81.29 91.90
下载: 导出CSV
表 4 方法应用
Table 4 Application of method
样品名称
sample name回收率/%
recovery平均回收率/%
average recovery标准偏差
SD相对标准偏差/%
RSD凡纳滨对虾
Penaeus vannamei88.67 88.94 3.59 4.0 92.66 85.49 牡蛎
Carnis ostreae91.71 90.69 1.88 2.1 91.83 88.52 三疣梭子蟹
Portunus trituberculatus84.81 82.82 2.22 2.7 83.23 80.42 鲤
Cyprinus carpio83.60 81.70 1.92 2.4 81.74 79.76 大菱鲆
Scophthalmus maximus94.95 90.54 3.82 4.2 88.14 88.53 鳜
Siniperca chuatsi87.91 84.02 3.39 4.0 82.39 81.75
下载: 导出CSV
-
[1] 张建勋, 李永才, 杨希国. 高效液相色谱法测定饲料中替米考星的含量[J]. 中国饲料, 2007(18): 38-39. doi: 10.3969/j.issn.1004-3314.2007.18.013 [2] 冯明教, 胡玉, 马满堂, 等. 兽药替米考星的研究进展[J]. 现代畜牧兽医, 2007(2): 44-45. doi: 10.3969/j.issn.1672-9692.2007.02.028 [3] 李雪红, 张煌涛, 占秀梅, 等. 动物性食品中大环内酯类抗生素残留分析综述[J]. 草食家畜, 2006(3): 10-12. doi: 10.3969/j.issn.1003-6377.2006.03.004 [4] 李存, 沈建忠, 江海洋. 猪组织中替米考星残留的高效液相色谱检测方法研究[J]. 畜牧兽医学报, 2005, 36(10): 1075-1078. doi: 10.3321/j.issn:0366-6964.2005.10.017 [5] 吴宁鹏, 周红霞, 郭芙蓉, 等. 牛奶中替米考星残留量的检测方法研究[J]. 中国兽药杂志, 2006, 40(12): 8-10. doi: 10.3969/j.issn.1002-1280.2006.12.003 [6] 闫春芝, 赵红彦, 秦朝英. 高效液相色谱法测定替米考星含量[J]. 中国兽药杂志, 2006, 40(8): 26-27. doi: 10.3969/j.issn.1002-1280.2006.08.009 [7] 芮萍. 绵羊血清中替米考星的高效液相色谱检测[J]. 河北科技师范学院学报, 2004, 18(3): 15-18. doi: 10.3969/j.issn.1672-7983.2004.03.005 [8] 张素梅, 杜向党, 吴宁鹏, 等. 牛肉中替米考星残留的高效液相色谱检测方法研究[J]. 黑龙江畜牧兽医, 2007(7): 82-84. doi: 10.3969/j.issn.1004-7034.2007.07.045 [9] 张旭东, 吕瑞娥, 张宝国. 替米考星的高效液相分析[J]. 郑州大学学报, 2003, 35(3): 70-72. doi: 10.3969/j.issn.1671-6841.2003.03.018 [10] 王敏, 林维宣, 郭德华, 等. 高效液相色谱-串联质谱法同时检测动物性食品中多种大环内酯类药物[J]. 分析测试学报, 2007, 26(5): 675-678. doi: 10.3969/j.issn.1004-4957.2007.05.018 [11] 陈莹, 陈辉, 林谷园. 超高效液相色谱串联质谱法对鳗鱼中大环内酯类、喹诺酮类和磺胺类兽药残留量的同时测定[J]. 分析测试学报, 2008, 27(5): 538-541. doi: 10.3969/j.issn.1004-4957.2008.05.021 [12] 赵东豪, 贺利民, 聂建荣. HPLC-MS/MS检测猪肉中六种大环内酯类抗生素[J]. 分析试验室, 2009, 28(1): 117-119. doi: 10.3969/j.issn.1000-0720.2009.01.030 [13] 王凤美, 陈军辉, 林黎明, 等. UPLC-MS/MS法对动物源性食品中12种大环内酯类抗生素残留的测定[J]. 分析测试学报, 2009, 28(7): 784-788. doi: 10.3969/j.issn.1004-4957.2009.07.006 [14] STOBBA-WUEY C M, CHANG J P, ELSBUTY D T, et al. Determination of tilmicosin residues in chicken, cattle, swine and sheep tissues by liquid chromatography[J]. J AOAC Inter, 2000, 83(4): 837-846. https://pubmed.ncbi.nlm.nih.gov/10995110/#:~:text=A%20method%20was%20developed%20and%20validated%20for%20determination,chicken%20fat%2C%20skin%2C%20and%20muscle%20over%20a%20concentrat
[15] 刘晔, 王洪新, 戴军, 等. 固相萃取-高效液相色谱法测定猪肝中的大环内酯类抗生素[J]. 食品与发酵工业, 2008, 34(5): 162-165. https://d.wanfangdata.com.cn/periodical/Ch9QZXJpb2RpY2FsQ0hJTmV3UzIwMjQxMTA1MTcxMzA0Eg5zcHlmeDIwMDgwNTAzNxoIanB4aHRzb2c%3D [16] DUBOIS M, FIUCHARD D, SIOR E, et al. Identification and quantification of five macrolide antibiotics in several tissues and milk by liquid chromatography-alectrospray tandem mass spectrometry[J]. J Chromatogr B, 2001, 753(2): 189-202. doi: 10.1016/S0378-4347(00)00542-9
[17] CODONY R, COMPANO R, GRANADOS M, et al. Residue analysis of macrolides in poultry muscle by liquid chromatography-electrospray mass spectrometry[J]. J Chromatogr A, 2002, 959(1/2): 131-141. doi: 10.1016/s0021-9673(02)00406-5
[18] HOFIE M, SAITO K, ISHII R, et al. Simultaneous determination of five macrolide antibiotics in meat by high-performance liquid chromatography[J]. J Chromatogr A, 1998, 812(1): 295-302. doi: 10.1016/S0021-9673(98)00004-1
[19] HORIE M, TAKEGAMI H, TOYAK, et al. Determination of spiramycin and tilmicosin in meat and fish by LC/MS[J]. Shokuhin Eiseigaku Zasshi, 2003, 44(3): 150-153. doi: 10.3358/shokueishi.44.150
[20] 徐锦忠, 吴宗贤, 杨雯筌, 等. 液相色谱-电喷雾串联质谱测定蜂蜜中8种大环内酯类药物残留[J]. 分析化学研究报告, 2007, 35(2): 166-170. doi: 10.3321/j.issn:0253-3820.2007.02.002 [21] 唐才明, 黄秋鑫, 余以义, 等. 高效液相色谱-串联质谱法对水环境中微量磺胺、大环内酯类抗生素、甲氧苄胺嘧啶与氯霉素的检测[J]. 分析测试学报, 2009, 28(8): 909-913. doi: 10.3969/j.issn.1004-4957.2009.08.007 [22] 王海涛, 张睿, 段宏安, 等. 高效液相色谱法同步检测牛奶中替米考星、泰乐菌素和螺旋霉素残留量[J]. 分析实验室, 2008, 27(7): 98-101. doi: 10.3969/j.issn.1000-0720.2008.07.027 -
期刊类型引用(2)
1. 黄珺,黄洪辉,吴风霞,戴明,齐占会. 嗜盐杆菌HSQAY1对中肋骨条藻的溶藻物质特性. 环境工程学报. 2016(04): 2077-2082 .  百度学术
百度学术
2. 魏玖红,娄永江,魏丹丹,宋美,方磊. 南美白对虾红变因素及控制技术研究. 食品工业科技. 2016(19): 148-152 . 百度学术
其他类型引用(1)








计量
- 文章访问数: 4693
- HTML全文浏览量: 158
- PDF下载量: 2690
- 被引次数: 3
 粤公网安备 44010502001741号
粤公网安备 44010502001741号
